New cyclic tetrameric and square-grid polymeric modes of supramolecular self-assembly of zinc tetra(4-pyridyl)porphyrin†
Sophia
Lipstman
and
Israel
Goldberg
*
School of Chemistry, Sackler Faculty of Exact Science, Tel Aviv University, 69978, Ramat Aviv, Tel Aviv, Israel. E-mail: goldberg@post.tau.ac.il; Fax: +972-36409293; Tel: +972-36409965
First published on 4th September 2009
Abstract
New supramolecular assembly patterns of zinc tetra(4-pyridyl)porphyrin into discrete (0D) cyclic tetrameric oligomers and polymeric 2D square-grids by self-coordination have been revealed.
The zinc tetra(4-pyridyl)porphyrin building block (ZnTPyP) was successfully used already in 1991 for the self-assembly of self-coordinated chain-oligomeric or 1D polymeric patterns.1 Our own efforts in the coordination chemistry of ZnTPyP resulted shortly thereafter in the synthesis and characterization of a unique single-framework 3D coordination polymer of this compound (in addition to 1D zig-zag polymeric aggregates).2 This framework solid is characterized by a robust honeycomb architecture of trigonal R
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) space group symmetry, exhibiting unprecedented thermal stability to >400 °C. The same structure, perforated by 0.5–0.6 nm wide channels, was found in a number of analogous porphyrin solids, revealing genuine porosity and remarkable sorption and desorption features.3,4 ZnTPyP has a planar and rigid structure and is prone to self-coordinate via the complementary metal center and peripheral 4-pyridyl sites. The difference between the above 1D and 3D (ZnTPyP)∞ arrays is in the number of binding sites used for coordination by the porphyrin scaffold. In the 1D polymers a given porphyrin unit is involved in two coordination bonds to its neighbors.1,2 One out of the four peripheral pyridyl substituents links to the zinc center of an adjacent species, while the five-coordinate metal ion binds to a pyridyl group of a third porphyrin unit. In the formation of the 3D R framework polymer (the occurrence of which has been reported repeatedly)2–6 the ZnTPyP molecule behaves as a tetradentate entity. The zinc center is six-coordinate ligating trans-axially to two pyridyl units of two different molecules. In addition, two trans-related pyridyl substituents of a given porphyrin bind to the metal centers of two other adjacent units. Association of one porphyrin to more than four porphyrin neighbors, or involvement of cis-related pyridyl groups in coordination to two adjacent ZnTPyP's appears to be unlikely for steric reasons. This leaves two trans-related pyridyls in every porphyrin molecule pointing away from the coordination sites and being exposed to solvation. The flexibility in the coordination environment of the zinc ion in ZnTPyP, allowing axial binding of either one or two ligands gave rise also to formation of ladder- and branched chain-type coordination polymers in which the two (5-coordinate square pyramidal and 6-coordinate octahedral) coordination modes co-exist within the same assembly.6,7 The above observations attest to the high propensity of the ZnTPyP moiety to exhibit diverse modes of self-coordination (also referred to as “supramolecular isomerism”).8 Application of ZnTPyP and related metalloporphyrins to the construction of polymeric networks with the aid of exocyclic metal ion connectors, has been widely studied,9 but these are out of scope of the present report.
space group symmetry, exhibiting unprecedented thermal stability to >400 °C. The same structure, perforated by 0.5–0.6 nm wide channels, was found in a number of analogous porphyrin solids, revealing genuine porosity and remarkable sorption and desorption features.3,4 ZnTPyP has a planar and rigid structure and is prone to self-coordinate via the complementary metal center and peripheral 4-pyridyl sites. The difference between the above 1D and 3D (ZnTPyP)∞ arrays is in the number of binding sites used for coordination by the porphyrin scaffold. In the 1D polymers a given porphyrin unit is involved in two coordination bonds to its neighbors.1,2 One out of the four peripheral pyridyl substituents links to the zinc center of an adjacent species, while the five-coordinate metal ion binds to a pyridyl group of a third porphyrin unit. In the formation of the 3D R framework polymer (the occurrence of which has been reported repeatedly)2–6 the ZnTPyP molecule behaves as a tetradentate entity. The zinc center is six-coordinate ligating trans-axially to two pyridyl units of two different molecules. In addition, two trans-related pyridyl substituents of a given porphyrin bind to the metal centers of two other adjacent units. Association of one porphyrin to more than four porphyrin neighbors, or involvement of cis-related pyridyl groups in coordination to two adjacent ZnTPyP's appears to be unlikely for steric reasons. This leaves two trans-related pyridyls in every porphyrin molecule pointing away from the coordination sites and being exposed to solvation. The flexibility in the coordination environment of the zinc ion in ZnTPyP, allowing axial binding of either one or two ligands gave rise also to formation of ladder- and branched chain-type coordination polymers in which the two (5-coordinate square pyramidal and 6-coordinate octahedral) coordination modes co-exist within the same assembly.6,7 The above observations attest to the high propensity of the ZnTPyP moiety to exhibit diverse modes of self-coordination (also referred to as “supramolecular isomerism”).8 Application of ZnTPyP and related metalloporphyrins to the construction of polymeric networks with the aid of exocyclic metal ion connectors, has been widely studied,9 but these are out of scope of the present report.
The occurrence of different crystalline coordination polymers is strongly affected by a combination of enthalpic (coordination strength), entropic (connectivity and dimensionality of the polymeric aggregate), solvation and product solubility factors, and often some of the possible structure modifications and coordination patterns cannot be isolated and remain on the drawing board. In the context of ZnTPyP, molecular modeling and experimental observations on analogous compounds indicated clearly that additional (to those already known—see above) supramolecular assembly modes should be feasible. For example, tetraaryl metalloporphyrins bearing only a single pyridyl function and three other substituents of considerable weaker coordination capacity, were shown to aggregate in the form of 0D discrete cyclic tetramers.10,11 The porphyrin units in the latter are interconnected to one another through two bonds per porphyrin only. The central zinc/metal ion is 5-coordinate, attracting the pyridyl group of an adjacent unit, while its pyridyl substituent binds to the metal center of a third porphyrin. The interporphyrin binding mode in these tetramers is the same as in the 1D polymeric assemblies of ZnTPyP,1,2 though the mutual orientation of neighboring porphyrins is different. Correspondingly, the appearance of a similar cyclic tetrameric oligomer of ZnTPyP (with three out of the four pyridyl groups not coordinating to the zinc ion) appeared to be only a question of time.
During our continuing search for new porphyrin-based coordination11 as well as hydrogen bonding12 networks, the 0D cyclic tetrameric oligomer of ZnTPyP (1) has been obtained by serendipity. The preparative procedure involved dissolving 0.007 mmol of ZnTPyP and 0.022 mmol of 2,5-thiophenedicarboxylic acid (TPDA) in a mixture of methanol and a minimal amount of o-chlorophenol. The resulting solution was then refluxed for 10 min to increase the solubility, and filtrated. X-Ray quality crystals of 1 were obtained by vapor diffusion method placing the filtrated reaction mixture for 4 days in a close vessel with diethyl ether.‡ The structure of the previously unobserved cyclic tetramer of ZnTPyP, which assembles around the tetragonal ![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) axis is illustrated in Fig. 1.
axis is illustrated in Fig. 1.
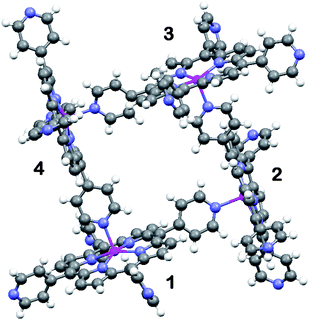 | ||
| Fig. 1 Ball-and stick illustration of the supramolecular (ZnTPyP)4 oligomer in 1. The individual porphyrin units are related to one another by the tetragonal symmetry axis (perpendicular to the projection shown, in the center of the tetramer). | ||
Within the tetrameric units the porphyrin entities are oriented nearly perpendicular to one another, forming a square coordination assembly with shortest H⋯H distances across the square box of 5.94 Å (which leaves a solvent accessible space of approximately 4 × 4 Å2). The 5-coordinate zinc ion deviates slightly towards the axial ligand at Zn–Npy = 2.140(3) Å (Zn–Npyrrole distances being within 2.06–2.08 Å). The tetrameric structure is very similar to that observed for the zinc and cobalt complexes of the mono(4-pyridyl)-tri(4-iodophenyl)-phorphyrins.11 The non-coordinated porphyrin fragments (exhibiting larger thermal displacement ellipsoids than the coordinated one) are solvated in the crystal by the o-chlorophenol solvent incorporated into the crystal lattice. Crystal packing of the tetramers in 1 is stabilized by dispersion.
It can be readily visualized from Fig. 1 that a slight change in the connectivity scheme can convert the coordination process from 0D tetrameric (as in 1) to 1D zig-zag polymeric (Fig. 2; as observed earlier).1,2 This would simply involve binding porphyrin-4 to the zinc center of porphyrin-1 from the lower side instead of from the upper side. The higher abundance of 1D ZnTPyP polymers, instead of the tetramers, may result from more severe geometric constraints associated with the formation of the latter. In the polymeric arrays the relative orientation of adjacent porphyrin rings is not restricted to be nearly perpendicular as in 1 (in fact, the preferred Zn(1)⋯Npy(2)⋯Zn(2) angle between two intercoordinated neighbors is approximately 155–160°),1,2 thus apparently lowering the activation energy for their formation.
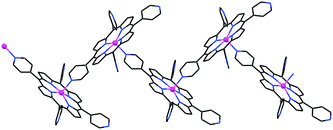 | ||
| Fig. 2 Wireframe illustration of the alternative 1D chain coordination mode via 2-bonds per porphyrin molecule, observed earlier.1,2 | ||
When a solution of 0.010 mmol TPDA in a mixture of methanol, DMF and water was carefully layered over a solution of 0.010 mmol ZnTPyP in a mixture of methanol and o-dichlorobenzene, single crystals of compound 2 were obtained after three days, which again didn't include the carboxylic acid moiety.‡ Characterization of the product shows that 2 represents a 2D mode of interporphyrin association of the ZnTPyP units, which involves coordination of every such unit to four neighboring ZnTPyP moieties. The zinc ion in the porphyrin core binds trans-axially to two pyridine N-atoms of two adjacent ZnTPyP molecules, while two trans-related pyridyl functions associate to the metal centers of two additional porphyrins (Fig. 3).
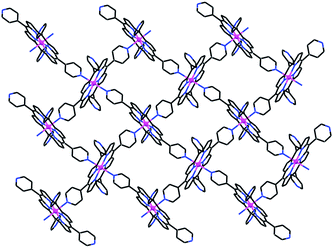 | ||
| Fig. 3 Illustration of the 2D “square-grid” coordination network of ZnTPyP in 2. The individual porphyrin units are located in the crystal on centers of inversion. Note the inclined approach of the axially ligating pyridyls to the corresponding zinc centers. | ||
The observed, somewhat distorted, square-grid porphyrin assembly consists of six-coordinate porphyrin complexes with equatorial Zn–Npyrrole and axial Zn–Npy bonds of 2.060–2.068(3) and 2.323(3) Å, respectively. It represents a supramolecular variant of the 3D honeycomb structure (shown in Fig. 4)2–5 with the same 4-bonds scheme of any given unit, but a different connectivity pattern within the intermolecular assembly. The mutual orientation of the neighboring molecules in the grid is not strictly perpendicular (the dihedral angle between the macrocyclic core rings of neighboring porphyrins is near 80°), due to the preferred inclined approach of the axially coordinated pyridyl group to the zinc center it associates with. Minimization of the interporphyrin void space within the layers may also contribute to the distortion of the grids from an ideally square geometry. The upper and lower surfaces of the 2D grids in 2 are lined with the two uncoordinated pyridyl substituents. In the crystal, the grids are offset-stacked along the a-axis, allowing the outward pyridyls of one layer to interdigitate between those of an adjacent layer. The layered arrays are held together by van der Waals forces. Correspondingly, this structure collapses upon removal of the crystallization solvent.
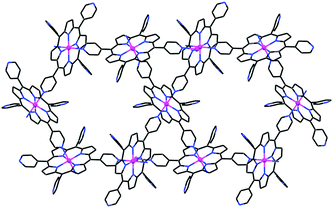
The topology of the coordination pattern of ZnTPyP in 2 is similar to the “paddle-and-wheel”-like assembly found only once before for the FeTPyP analog.13 Prior to our work this supramolecular binding pattern has not been reported for any other MTPyP compound, as opposed to the more abundant occurrence of the 3D modification (Fig. 4).2–5,14 The main topological difference between the 2D and 3D organizations lies in the relative orientations of the intercoordinated porphyrins, a degree of freedom which arises from the possible rotation of one framework with respect to the other about the intermolecular Zn–pyridyl bond. While in 2 neighboring ZnTPyP units are mutually nearly perpendicular (Fig. 3), this is not the case in the 3D assembly (Fig. 4), which gives rise to the topologically different organization. Evidently, the latter represents thermodynamically more robust architecture due to the more extended coordination pattern.
The ZnTPyP scaffold has one metal center, which can coordinate axially to either one or two ligands, and four divergent pyridyl ligating sites (of which at most only two can be used in the self-assembly process). These provide a considerable number of degrees of freedom to invoke different modes of supramolecular organization,2–7 which sometimes crystallize out concomitantly from a given reaction mixture. In a sense, the appearance of different modes of supramolecular self-assembly of a given compound has much in common with the occurrence of polymorphic intermolecular organizations in molecular crystals. Diverse interaction schemes are possible with the ZnTPyP, utilizing in a given product either only the square-pyramidal, the octahedral, or simultaneously both modules of intermolecular coordination. The number of new structural modifications (different networks formed from the same components)8 discovered is proportional to the time and effort spent to this end on the experimental bench. This report contributes two additional, long expected, self-coordinated forms of ZnTPyP to the continuously growing library of the structural variants with different connectivity schemes of this compound.
This research was supported by The Israel Science Foundation, Grant No. 502/08.
Notes and references
- E. B. Fleischer and A. M. Shachter, Inorg. Chem., 1991, 30, 3763 CrossRef CAS.
- H. Krupitsky, Z. Stein, I. Goldberg and C. E. Strouse, J. Inclusion Phenom. Mol. Recognit. Chem., 1994, 18, 177 CrossRef CAS.
- K.-J. Lin, Angew. Chem., Int. Ed., 1999, 38, 2730 CrossRef CAS.
- E. Dieters, V. Bulach and M. W. Hosseini, Chem. Commun., 2005, 3906 RSC.
- S. George and I. Goldberg, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, m1441 CrossRef.
- D. J. Ring, M. C. Aragoni, N. R. Champness and C. Wilson, CrystEngComm, 2005, 7, 621 RSC.
- Y. Diskin-Posner, G. K. Patra and I. Goldberg, J. Chem. Soc., Dalton Trans., 2001, 2775 RSC.
- B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629 CrossRef CAS.
- Representative references of TPyP coordination networks through exocyclic metal ions: D. Hagrman, P. J. Hagrman and J. Jubieta, Angew. Chem., Int. Ed., 1999, 38, 3165 Search PubMed; C.V.K. Sharma, G.A. Broker, J.G. Huddleston, J.W. Baldwin, R.M. Metzger and R.D. Rogers, J. Am. Chem. Soc., 1999, 121, 1137 CrossRef CAS; L. Carlucci, G. Ciani, D. M. Proserpio and F. Porta, Angew. Chem., Int. Ed., 2003, 42, 317 CrossRef CAS; T. Omura, A. Usuki, K. Fukumori, T. Otha and K. Tatsumi, Inorg. Chem., 2006, 45, 7988 CrossRef CAS.
- M. Vinodu, Z. Stein and I. Goldberg, Inorg. Chem., 2004, 43, 7582 CrossRef CAS.
- S. Lipstman, S. Muniappan and I. Goldberg, Cryst. Growth Des., 2008, 8, 1682 CrossRef.
- R. Koner and I. Goldberg, CrystEngComm, 2009, 11, 1217 RSC.
- L. Pan, S. Kelly, X. Huang and J. Li, Chem. Commun., 2002, 2334 RSC.
- A more detailed comparison between the 3D and 2D supramolecular assemblies of ZnTPyP is given in: R. Koner and I. Goldberg, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2009, 65, m139 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
Footnotes |
| † CCDC reference numbers 741066 and 741067. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b914799c |
| ‡ Crystal data:1, [C40H24N8Zn] (excluding a severely disordered molecule of o-chlorophenol solvent): formula weight 682.04, tetragonal, space group P 42/n, a = 23.5744(3), b = 23.5744(3), c = 15.0025(3) Å, V = 8337.7(2) Å3, Z = 8, T = 110(2) K, Dcalc = 1.087 g cm−3, 43170 collected data and 9932 unique reflections (θmax = 27.91°), Rint = 0.095. After “Squeeze”, the final R1 = 0.066 for 4843 observations with Fo > 4σ(Fo), R1 = 0.132 (wR2 = 0.187) for all unique data, |Δρ| ≤ 0.41 e Å−3. The o-chlorophenol moiety of the asymmetric unit was clearly identified in the electron-density maps, but it couldn't be reliably modeled by discrete atoms due to severe positional and orientational disorder. Its contribution was subtracted from the diffraction pattern by the “Squeeze” method.15 2, 1, [C40H24N8Zn] (excluding molecules of unidentified and disordered solvent): formula weight 682.04, monoclinic, space group C2/c, a = 20.8956(7), b = 13.6507(5), c = 14.6756(7) Å, β = 106. 202(1)°, V = 4019.8(3) Å3, Z = 4, T = 110(2) K, Dcalc = 1.127 g cm−3, 18471 collected data and 4671 unique reflections (θmax = 27.86°), Rint = 0.092. After “Squeeze” the final R1 = 0.075 for 2748 observations with Fo > 4σ(Fo), R1 = 0.127 (wR2 = 0.208) for all unique data, |Δρ| ≤ 0.53 e Å−3. The crystal structure contains unidentified solvent (probably a mixture of methanol and DMF), which could not be modeled by discrete atoms. Its contribution was subtracted from the diffraction pattern by the “Squeeze” method.15 The uncoordinated pyridyl substituents also reveal rotational disorder. The uniform identity of the crystalline materials was confirmed in each case by repeated measurements of the unit-cell dimensions from different single crystallites. |
| This journal is © The Royal Society of Chemistry 2010 |