Surprisingly strong effect of stabilizer on the properties of Au nanoparticles and Pt^Au nanostructures in electrocatalysis†
Gui-Rong
Zhang
and
Bo-Qing
Xu
*
Innovative Catalysis Program, Key Lab of Organic Optoelectronics & Molecular Engineering, Department of Chemistry, Tsinghua University, Beijing, 100084, China. E-mail: bqxu@mail.tsinghua.edu.cn; Fax: (+86)10-6279-2122
First published on 11th October 2010
Abstract
We show that the electrocatalytic properties of nearly monodispersed Au nanoparticles (NPs) and their derived Pt-on-Au (Pt^Au) nanostructures with similarly dispersed Pt entities are dependent on the nature of stabilizers involved in the colloidal syntheses of the Au particles. The effect of stabilizer on the activity for oxygen reduction reaction (ORR) of Au NPs significantly outweighed the Au nano-size effect and thus would raise an alert to those reported size-dependent properties of metal NPs carrying various stabilizers in earlier studies. It is also demonstrated that the stabilizer effect on the property of Au NPs can further induce changes in the catalytic properties of their carried Pt. These findings clearly suggest that a proper screening of the stabilizer in the colloidal synthesis of metal NPs would be important for innovative nanomaterials and catalysts.
Introduction
Nanoscale particles or nanoparticles (NPs) of metals have distinguished themselves from their bulk counterparts with unique chemical and electronic properties,1–3 which are promising for many important applications in optoelectronic and magnetic devices, thermal- and electro-catalysis, chemical- and bio-sensors.4–6 A very fundamental aspect in this field is to understand how the properties of a particle for a specific application would change with its particle size and/or shape. This relies at least in experimental studies on the availability of size-controlled synthesis methods. The colloidal synthesis method has the advantage of precisely tailoring the metal particle sizes and/or shapes and their distributions usually by varying the type or dosage of a stabilizer.7–9 However, there is always the possibility that an alternation of the stabilizer in the syntheses would significantly affect the size- and/or shape-dependent properties of the metal NPs. Many publications addressed the size-dependent properties of metal colloids, but few of them actually concerned the influence from stabilizers.9–12Due to the complicated nature of the metal sizes and shapes in either supported or unsupported heterogeneous catalysts, the understanding of size- and shape-dependent catalysis of metal NPs has been a long endeavor of catalysis research. A possible disturbance by the ‘residual’ stabilizers of their size- and/or shape-dependent catalysis could not be excluded as long as metal NPs were derived from colloidal syntheses.13,14 In the search for advanced electrocatalysts, our previous study revealed that small Pt flecks with higher than 40% dispersions (utilizations15,16) on ca. 3 nm Au NPs prepared with poly(vinylalcohol) (PVA) as the stabilizer showed dramatically higher activity for formic acid (HCOOH) electrooxidation than their counterpart Pt deposits on ca. 10 nm Au NPs with citrate as the stabilizer, which uncovers a size effect of Au NPs on their carrying Pt without considering a possible function of the stabilizers.9 Similarly, strong size-dependent activity of gold for cathode ORR was recorded on differently sized Au NPs prepared with varied reductants and stabilizers.12 It could be possible that these observed size effects of Au NPs9,12 can actually be a consequence of combined actions of the Au particle size and the stabilizer.
Attempts were already made recently in a few publications to show the possible effect of the stabilizer on the catalytic properties of metal NPs.17–19 In these publications, however, variation in the stabilizers was usually accompanied by changes in the metal particle sizes which could shake the basic requirement for similarly sized metal NPs. A very recent work investigated the effect of stabilizer on Pt NPs for CO oxidation and ethylene hydrogenation catalysis, in which three different stabilizers poly(vinylpyrrolidone) (PVP), trimethyl tetradecyl ammonium bromide (TTAB) and oleylamine (OA) were adsorbed onto the pre-formed Pt NPs from a polyol reduction protocol.20 The addition of PVP was shown to produce the highest catalytic activity. Nevertheless, in a practical colloidal synthesis, a stabilizer is usually added before enabling the reduction of the metal precursor. The presence of the stabilizer during reduction would, however, influence not only the particle size and/or shape but also the internal crystal structure of the as-formed metal NPs, which are usually strongly correlated to their catalytic properties.21 Therefore, the post-added stabilizer effects20 may not be relevant to those involved in a practical colloidal method.
Herein, we report our assessment on the influence of PVP, PVA and citrate stabilizers on the electrocatalytic properties of similarly sized Au NPs and their derived Pt^Au nanostructures9,16,22,23 by direct syntheses of similarly sized Au NPs with average diameters at 3, 5 and 10 nm, respectively, in the presence of the individual stabilizers. The Au NPs were prepared via colloidal methods including conventional colloidal syntheses using either PVA or citrate as the stabilizer and, alternatively, a seed-mediated growth method using PVP as the stabilizer. The samples were denoted as Au-d-S, where d and S refer to the average diameter in nanometres of Au NPs and the name of the stabilizer, respectively. These Au NPs were employed as substrate to disperse Pt entities by reductive deposition of Pt at the atomic ratio Pt/Au = 0.10, which generated Pt^Au nanostructures (coded as Pt0.10^Au-d-S) as in our earlier studies.9,16,22,23 We have found that the mass-specific activity (MSA, current density normalized to the loading of metal) of Au for the cathode ORR in Au-d-PVA and -Citr is enhanced to 5∼6 times that in Au-d-PVP when their Au particles were comparably sized. Furthermore, we uncover that the catalytic properties of Pt in Pt^Au nanostructures are also significantly affected by the variation of the Au stabilizer; the Pt MSA and areal/intrinsic activity (IA, current density normalized to the active surface area of Pt) for HCOOH electrooxidation in Pt0.10^Au-d-PVP are enhanced to 2∼3 times that in Pt0.10^Au-d-PVA and -Citr. These findings clearly reveal that the stabilizer molecules actually impose pronounced effects on the catalytic properties of their stabilized metal NPs. Therefore, a proper screening of the stabilizer on metal NPs would also be crucial for creating more efficient metal catalysts and innovative nanomaterials.
Experimental
Au-d-PVP (d = 3, 5, 10) were prepared via a seed-mediated growth method. The primary seeds with an average diameter around 1.9 nm were prepared according to Tsukuda's method:24 555 mg PVP (MW = 10000) was added to 50 mL aqueous solution of HAuCl4 with a concentration of 1.0 × 10−3 mol L−1, and subsequently an ice-cooled solution of NaBH4 was quickly injected into the solution to make the primary Au seeds, followed by a stepwise growth approach to synthesize Au particles with a desired mean particle size. Specifically, a certain amount of Au seeds were added to 35 mL aqueous HAuCl4 solution containing 555 mg PVP, while the solution was stirred, 15 mL of freshly prepared L-ascorbic acid (5.0 × 10−3 mol L−1) was added dropwise (3 mL min−1) into the above mixed solution from a buret, and the detailed preparation conditions were shown in Table S1 of the ESI† and ref. 25. The citrate- and PVA-stabilized Au NPs (Au-10-Citr, Au-3-PVA, Au-5-PVA) were prepared according to procedures reported previously.16,22Pt0.10^Au-d-S nanostructures were synthesized by hydrogen-reduction of PtCl62− onto the pre-formed Au NPs at a fixed atomic ratio Pt/Au = 0.10 according to the protocol described previously.16
The carbon-supported Au-d-S and Pt0.10^Au-d-S nanostructures were prepared by mixing a desired amount of Vulcan XC-72 carbon black (BET surface area: 240 m2 g−1) with Au-d-S or Pt0.10^Au-d-S hydrosols, followed by careful adjustment of the solution acidity to pH = 1.5 with 1 mol L−1 HNO3. Then the suspension was refluxed for 2 h under vigorous stirring. The solid was separated using filtration, followed by intensive washing with deionized water and air-drying at 110 °C for 2 h to give the Au-d-S/C or Pt0.10^Au-d-S/C samples. The loading of Au was controlled at 5 wt% in all of the Au-d-S/C and Pt0.10^Au-d-S/C samples.
Transmission electron microscopy (TEM) images were captured using a JEOL JEM-2010 microscope operated at 120 kV. High-resolution TEM images were obtained on Philips CM200 FEG or Tecnai F20 (both at 200 kV accelerating voltage). UV-Vis spectroscopy was recorded on a Unico UV-2102PC spectrometer with a resolution of 0.5 nm. X-ray photoelectron spectroscopy (XPS) measurements were carried out on a PHI 5300 ESCA1610 SAM instrument equipped with Mg Kα radiation. The actual loading amount and composition of Pt and Au in the as-prepared carbon-supported catalysts were determined by inductively coupled plasma atomic emission spectrometry (ICP-AES, IRIS Intrepid II XSP, ThermoFisher), and the loading of Au was determined to be around 5 wt% in all the investigated Au-d-S/C and Pt0.10^Au-d-S/C samples.
Electrochemical measurements were performed in a three-electrode electrochemical cell using a potentiostat/galvanostat model 263A (PAR) operated by PowerSuite software. A saturated calomel electrode (SCE) and a 1.0 cm × 1.0 cm Pt foil were used as reference and counter electrodes, respectively. The potentials reported here are given relative to SCE. The working electrode was prepared using the procedure reported previously.16 HCOOH electrooxidation study was done using cyclic voltammetry (CV) in 0.5 M H2SO4 with 2.0 M HCOOH in the potential range from −0.2 to 1.0 V (scan rate: 20 mV s−1). Measurement of the working electrode for ORR was performed in O2-saturated 0.5 M KOH solution using a glass carbon rotating disk electrode at a rotation rate of 1600 rpm (scan rate: 10 mV s−1).
Results and discussion
Fig. 1 shows the representative TEM images for some of the as-prepared Au NPs (see also Fig. S1 in the ESI†). The PVP-stabilized Au NPs were obtained with size distribution at 3.2 ± 0.5, 4.7 ± 0.7 and 10.3 ± 1.8 nm, respectively; the PVA-stabilized particles were obtained at 3.0 ± 0.6 and 5.0 ± 1.5 nm, and the citrate-stabilized ones at 10.0 ± 1.2 nm (Table 1). The high resolution particle morphologies for every Au-3-S, Au-5-S and Au-10-S samples were dominated by fivefold twinned particle structures in icosahedron and decahedron shapes, as shown by the particles probed with HRTEM in the insets of Fig. 1. Thus, the present variation in the stabilizers didn't cause notable dissimilarity in the internal crystal structure of Au NPs.| Sample | d Au /nm | EASb/(m2 gPt−1) | U Pt /(%) | IA/(A mPt−2) | MSA/(A mgPt−1) |
|---|---|---|---|---|---|
| a d Au is the average size of Au particles. More than 300 randomly selected particles were measured for size and distribution analysis. b EAS is obtained by measuring the charges associated with hydrogen desorption signals on the CV curves in 0.5 M H2SO4. | |||||
| Pt^Au-3-PVP/C | 3.2 ± 0.5 | 209.8 | 88.5 | 28.1 | 5.8 |
| Pt^Au -5-PVP/C | 4.7 ± 0.7 | 174.5 | 73.6 | 25.0 | 4.4 |
| Pt^Au-10-PVP/C | 10.3 ± 1.8 | 136.7 | 57.7 | 13.1 | 1.8 |
| Pt^Au-3-PVA/C | 3.0 ± 0.6 | 218.5 | 90.4 | 14.1 | 3.1 |
| Pt^Au-5-PVA/C | 5.0 ± 1.5 | 183.5 | 77.4 | 14.0 | 2.6 |
| Pt^Au-10-Citr/C | 10.0 ± 1.2 | 126.0 | 53.4 | 4.3 | 0.5 |
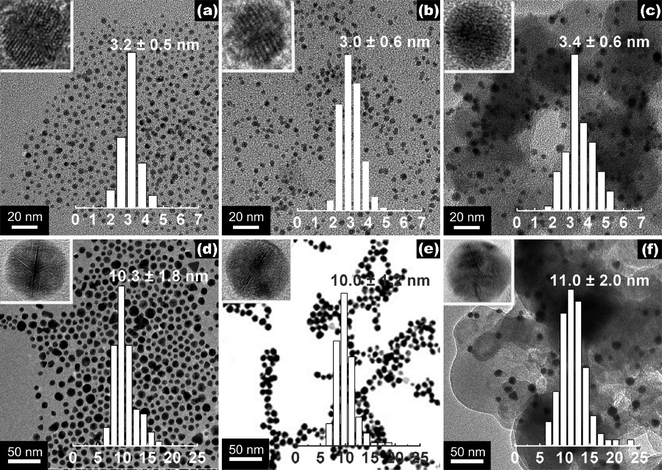 | ||
| Fig. 1 Representative TEM images and size histograms of (a) Au-3-PVP, (b) Au-3-PVA, (c) Au-3-PVP/C, (d) Au-10-PVP, (e) Au-10-Citr and (f) Au-10-PVP/C. The insets are the representative HRTEM images of the corresponding single particle, showing icosahedral structures. | ||
Au NPs could serve as electrocatalysts for the cathode ORR.12 The present similarly sized Au NPs carrying varied stabilizers, were loaded onto Vulcan XC-72 carbon black to make carbon-supported Au NPs catalysts (Au-d-S/C) for measuring their activity as cathode ORR catalysts in alkaline electrolyte. As confirmed with TEM measurements (e.g., Fig. 1c & f), Au NPs retained their sizes and narrow distributions after they were loaded onto the carbon support.
Fig. 2a shows the polarization curves of ORR on these Au-d-S/C catalysts in the alkaline electrolyte. The half-wave potential (E1/2) and kinetic current, two important parameters in the quantitative assessment of an electrocatalyst for a given electrochemical reaction, varied sensitively with the variation in nature of the Au stabilizers for those samples having similarly sized Au NPs for the catalysis. For instance, the E1/2 of ORR on Au-3-PVA/C (−0.17 V) shifted positively by ca. 70 mV compared with the number on Au-3-PVP/C (−0.24 V). Fig. 2b correlates the normalized mass-transport corrected kinetic currents at −0.20 V, that is MSA of Au, with the Au particle sizes. The Au MSA in Au-3-PVA/C was 296.6 mA mgAu−1, which is 6 times that in Au-3-PVP/C (44.6 mA mgAu−1). The E1/2 and Au MSA for Au-10-Citr/C were ca −0.17 V and 154.5 mA mgAu−1, respectively; both parameters are remarkably higher than its PVP-stabilized counterpart catalyst (Au-10-PVP/C), whose numbers were E1/2 = −0.25 V and Au MSA = 23.1 mA mgAu−1. Apparently, Au NPs stabilized by PVA and citrate exhibited much higher catalytic activity for ORR than those stabilized by PVP. The data obtained on the other two samples having an average Au particle size at 5 nm (i.e., Au NPs in Au-5-PVA/C and Au-5-PVP/C) also demonstrated the inferiority of the PVP-stabilized Au NPs for ORR. This is to the best of our knowledge the first time to clearly demonstrate, on the basis of similar particle sizes and internal crystal structures, a dramatic effect of the colloid stabilizer molecules on the electrocatalytic properties of metal NPs.
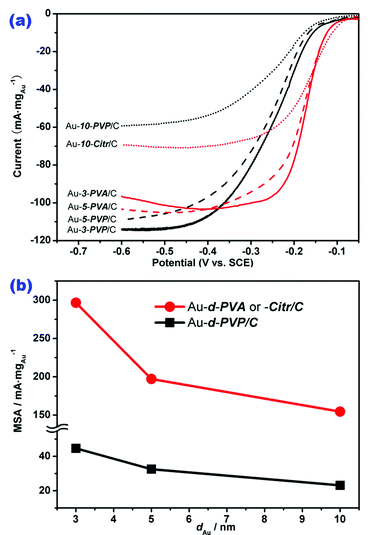 | ||
| Fig. 2 (a) Polarization curves for ORR on carbon-supported Au NPs (current normalized to the loading of Au). (b) Plots of Au MSA for ORR as a function of particle size of Au. | ||
It is surprising that the Au nano-size effect indicated by each of the individual data line in Fig. 2b is much lower than the difference between the two data lines, showing that the effect of PVP stabilizer (or capping agent) on the catalysis of Au NPs for ORR significantly outweighed the effect of Au particle size. This is very memorable as totally confusing conclusions would have been reached if the size-dependent activity of Au NPs was investigated without considering the nature of their involved stabilizer molecules. Therefore, we have a reason to raise an alert to those reported size-dependent properties of metal NPs carrying totally different stabilizers in the earlier literature.
On having Pt dispersed as small islands and/or flecks of varying dispersions on Au and Ag NPs from colloidal syntheses, recent studies in this laboratory have presented several series of nanostructured Pt-on-M (Pt^M, M being Au or Ag) catalysts for the electrooxidation of methanol and HCOOH.9,16,22,23,26,27 Two series of Pt^Au samples made from Au-10-Citr and Au-3-PVA (named previously Pt^Au–I and II), respectively, showed dramatically different Pt activity for the electrooxidation of HCOOH; on the basis of similar Pt dispersions, the activity of Pt on Au-3-PVA (Pt^Au-II) was always more than twice that on Au-10-Citr (Pt^Au–I), which was attributed solely to the size effect of Au.9 That size effect of Au on Pt^Au for the anode HCOOH electrooxidation was revisited below with the present understanding of the stabilizer effect on Au NPS discussed above, that is by measuring the anode activity of Pt0.10^Au-d-S for HCOOH electrooxidation.
The formation of Pt0.10^Au-d-S nanostructures by reductive deposition of Pt from PtCl62− ions on the Au-d-S NPs was characterized by UV-Vis spectroscopy (Fig. 3). Compared with their Au-d-S counterparts, the weakened but still clearly observable Au surface plasmon absorptions from the Pt0.10^Au-d-S samples revealed that Pt entities loaded on the surface of Au NPs at the atomic ratio Pt/Au = 0.10 were not sufficient to completely cover the underlying Au surfaces.22 The TEM images of Pt0.10^Au-d-S (Fig. S1 in the ESI†) were found indistinguishable from those of Au-d-S, probably due to the low Pt/Au ratio. These Pt0.10^Au-d-S particles were then loaded onto Vulcan XC-72 carbon black to make Pt0.10^Au-d-S/C for electrocatalysis study. It was confirmed that the loading of these Pt0.10^Au-d-S particles on the carbon support had no detectable effect on the metal particle sizes, as evidenced in Fig. S1 of the ESI†.
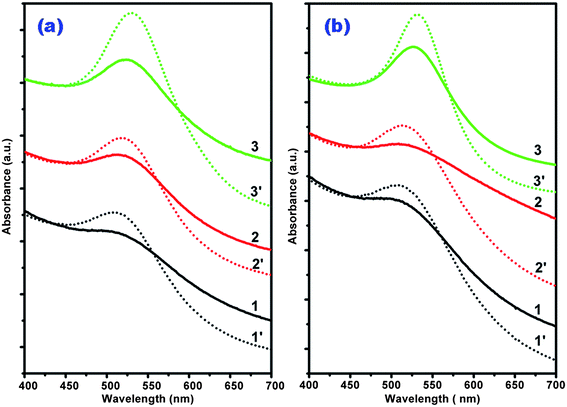 | ||
| Fig. 3 UV-Vis spectra of the as-prepared Au NPs (dotted lines) and Pt0.10^Au (solid lines) nanostructures. (a) 1. Pt0.10^Au-3-PVP; 2. Pt0.10^Au-5-PVP; 3. Pt0.10^Au-d-PVP. (b) 1. Pt0.10^Au-3-PVA; 2. Pt0.10^Au-5-PVA; 3. Pt0.10^Au-10-Citr. | ||
Table 1 gives the electrochemically active surface area (EAS) and Pt utilization (UPt)15 in every Pt0.10^Au-d-S sample. It deserves to be noted that the EAS and UPt data are very close for those Pt0.10^Au-d-S samples having a fixed d number (or with similar Au sizes). This observation indicates that the stabilizers used in this study imposed little effect on the exposed percentage (dispersion) of Pt or on charges associated with the hydrogen adsorption and desorption at the Pt surface on the CV curves. Consequently, two resembling decreasing trends with an increasing average Au particle size of UPt in the Pt0.10^Au-d-S samples were clearly revealed in Table 1, e.g., UPt in Pt0.10^Au-3 was ca. 90%, nearly twice that in Pt0.10^Au-10. This is not surprising since for a given amount of Au, the smaller particle would provide a larger surface area to disperse Pt.22 Thus, the Pt deposits exist as highly dispersed two-dimensional islands or flecks at the surface of Au NPs (see Scheme 1) with UPt higher than 50%; the domain size of Pt in Pt0.10^Au-d-S increased with the increase in d, as demonstrated in our earlier papers.9,16,22
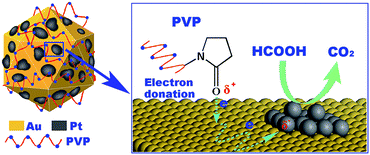 | ||
| Scheme 1 Dispersion state of Pt on PVP-stabilized icosahedral Au NPs (Pt0.10^Au-d-PVP); the Pt flecks become electron-rich due to PVP–Au interaction. | ||
Fig. 4 shows the anodic-scan CV curves for HCOOH electrooxidation on Pt0.10^Au-d-S/C (complete CV curves are shown in Fig. S2 of the ESI†). Apparently, all of the Pt0.10^Au-d-S/C catalysts give current peaks at potentials lower than 0.6 V, which suggests that HCOOH electrooxidation over these catalysts proceeded predominantly through a dehydrogenation mechanism or by the direct reaction pathway.9 In other words, the dehydration pathway leading to the formation of at least one poisonous surface intermediate like COads, is basically suppressed on these Pt^Au nanostructures irrespective of the stabilizer. On the backward scans (cathodic-scan curves, see Fig. S2 of the ESI†), the current peak developed most sharply on the Pt0.10^Au-3-S/C catalysts and the slope declined with the increase in the Au sizes (d) in the Pt0.10^Au-d-S/C samples, which may signify that the flecks with lower UPt or on larger Au NPs are less active for the electrooxidation of HCOOH.9
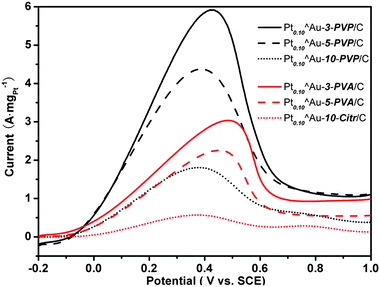 | ||
| Fig. 4 Anodic-scan curves of CV for HCOOH electrooxidation on carbon supported Pt^Au nanostructures. | ||
The MSA and IA of Pt according to the anodic current at 0.4 V are compared in Table 1. These activity data were also affected strongly by the stabilizer molecules (S) involved in the synthesis of Au NPs. The data for Pt0.10^Au-3-PVP/C were 5.8 A mgPt−1 and an IA of 28.8 A mPt−2, which were significantly higher than the ones for Pt0.10^Au-3-PVA/C (3.1 A mgPt−1 and 14.1 A mPt−2). Similarly, the activity data for Pt0.10^Au-5-PVP/C and Pt0.10^Au-10-PVP/C catalysts were also remarkably higher than those for Pt0.10^Au-5-PVA/C and Pt0.10^Au-10-Citr/C, respectively; the data for Pt0.10^Au-10-PVP/C were even 3 times that for Pt0.10^Au-10-Citr/C.
We confirmed in separate experiments that the Au-d-S particles without Pt were totally inactive for HCOOH electrooxidation, as shown previously for Au-3-PVA and -10-Citr in ref. 9. The significant activity differences of Pt in Pt0.10^Au-d-S with similar Au sizes (d) may therefore arise from an influence of the stabilizer molecules (S) on the interaction between Pt flecks and their underlying Au NPs, which again demonstrates that the stabilizers involved in the colloidal synthesis do affect the properties of the Au NP product. Great endeavors have hitherto been made to investigate the interaction between the stabilizer and their stabilized metal NPs.28–33 The adsorbed stabilizer could geometrically encapsulate the metal NPs. However, organic stabilizers on metal surface are usually in loose and open structures, and there would be abundant channels that keep the metal surface accessible to small molecules (like HCOOH) for adsorption and reaction.34
Somorjai et al. showed that the Pt surface would not be blocked by PVP molecules unless the sample was pretreated at high temperatures that could lead to a decomposition of PVP (e.g., 170 °C in reducing atmosphere).20 There was evidence that polymer stabilizers (PVP and PVA) could stabilize the metal NPs with multiple interaction sites in the fashions that permit the metal NPs retain most of their inherent surface activity.28,33 In the present study, the nearly invariant Pt EAS numbers for Pt0.10^Au-d-S/C catalysts with similarly sized Au NPs (i.e., similar d sizes, Table 1) imply that the stabilizer variations have no significant influence on the accessibility to the Pt sites. As is shown in Fig. 2, the appreciable catalytic activity for ORR of Au NPs carried out with various stabilizers (PVP, PVA or citrate) hints that the surfaces of such “protected” Au NPs are also accessible to O2 molecules. Scheme 1 gives a rough image for understanding the relationship between the stabilizer molecules and their protected Au NPs in Pt0.10^Au-d-PVP samples. However, a simple geometric effect is not sufficient to rationalize the remarkable stabilizer-dependent catalytic properties observed here in this study for ORR and HCOOH oxidation.
The hybrid DFT calculations of Haruta et al. indicated that the interaction of PVP with Au13 clusters would induce a change in the Au electronic structure due to electron donation from PVP to Au.29 This was later confirmed experimentally by Tsukuda et al. whose X-ray photoelectron spectroscopy (XPS), infrared (IR) and X-ray absorption near edge structure (XANES) measurements showed that the PVP-stabilized Au NPs (1∼2 nm) were negatively charged due to the electron donation from PVP.28 Electron donation from PVP to other metal NPs, e.g., Pt and Ag NPs, was also documented.31,32 As shown in Fig. 5, we found that the XPS binding energies of Au 4f electrons were lowered by ca. 0.9 eV for the Au-d-PVP particles relative to those for bulk Au, but the Au 4f binding energies for the Au-d-PVA and -Citr were found to be the same as for bulk Au. Therefore, the Au-d-PVP particles could be negatively charged in a sense that they were donated electrons from the stabilizing PVP molecules, compared with the Au-d-PVA and -Citr particles.
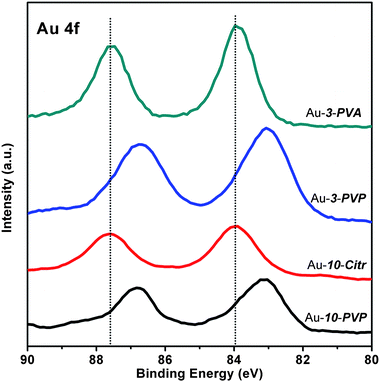 | ||
| Fig. 5 XPS spectra (Au 4f) of Au-3-S and Au-10-S particles. The vertical dotted lines mark the binding energy positions for bulk gold. | ||
The negatively charged Au NPs in Au-d-PVP would be responsible for their significantly lower catalytic activity for the cathode ORR (Fig. 2). Although the negatively charged Au particles would favor O2 adsorption according to recent calculations,35,36 the adsorption on Au-d-PVP might be too strong to allow the surface for further reaction.37
Our catalytic data for HCOOH electrooxidation over Pt0.10^Au-d-S/C (Table 1) demonstrate that the stabilizer effect on the electronic structure of Au NPs can further induce change in the catalytic properties of their carried Pt. It would be possible that those highly dispersed Pt flecks in the Pt0.10^Au-d-PVP could become electron-rich on interaction with the negatively charged PVP-stabilized Au NPs (Scheme 1), and thus promote the formation of adsorbed formate,38 a key surface intermediate regarded as the product of the rate determining step for HCOOH oxidation on Pt surface.39 The increased electron density in Pt would be beneficial to HCOOH activation due to preferred electron back-donation from Pt to the 2π* orbital of the adsorbate,40 which also accounts for the higher activity of Pt in Pt0.10^Au-d-PVP than that in Pt0.10^Au-d-PVA and -Citr samples.
The MSA and IA data of Pt in Pt0.10^Au-d-S/C were correlated with the sizes of Au NPs (d) in Fig. 6. It is apparent that the smaller the Au NPs the higher the catalytic activity both in terms of Pt MSA and IA, which confirms our earlier observation on the Ptm^Au-10-Citr/C and Ptm^Au-3-PVA/C samples.9 These Au nano-size effects on the activity of their carried Pt were qualitatively independent of the nature of the stabilizers. The correlations based on Pt0.10^Au-d-PVP/C catalysts (the squares) provide a relatively clear Au nano-size effect because the catalytic Pt0.10^Au nanostructures are concerned with the same stabilizer (PVP). The increment in Pt MSA with reducing the Au sizes for Pt0.10^Au nanostructures would be expected since smaller Au particles mean a higher gold surface area which would result in higher Pt utilization (dispersion).22 The IA numbers of Pt in Pt0.10^Au-5-S/C and -3-S/C were more or less similar but were doubled when compared to those in Pt0.10^Au-10-S/C samples (see also Table 1), which basically agree with our earlier data that IA of Pt in Pt^Au nanostructures was independent of Pt dispersion at UPt ≥ 40%.9 These data seem to suggest that Au particles smaller than ca. 5 nm could serve as some kind of “activity promoter” for the highly dispersed Pt flecks.9 Further study is needed to clearly identify this point.
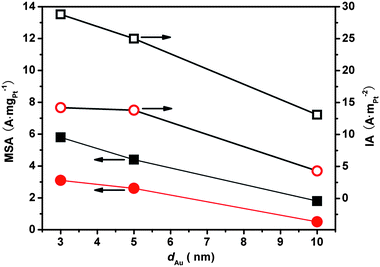 | ||
| Fig. 6 Mass-specific (solid data points) and intrinsic activity (empty data points) of Pt as a function of particle size of Au in Pt0.10^Au-d-PVP/C (squares), Pt0.10^Au-d-PVA/C and –Citr/C (circles) for HCOOH electrooxidation at 0.4 V. | ||
Conclusions
In conclusion, electrocatalytic properties of Au NPs and Pt^Au nanostructures were found dependent on the nature of stabilizers involved in the colloidal syntheses of the Au particles. The effect of stabilizer on the activity for ORR of Au significantly outweighed the Au nano-size effect. This finding would raise an alert to those reported size-dependent properties of metal NPs carried out with various stabilizers in the earlier literature. It is also demonstrated that the stabilizer effect on the properties of Au NPs can further induce changes in the catalytic properties of their carrying Pt in Pt^Au nanostructures. Thus, a proper screening of the stabilizer in colloidal synthesis of metal NPs would be important for innovative nanomaterials and catalysts.Acknowledgements
The authors wish to thank Dr Dang-Sheng Su (Fritz-Haber-Institut der Max-Planck-Gesellschaft, Berlin, Germany) and Dr Gang Liu (National Center for Nanoscience and Technology, Beijing, China) for their kind help in HRTEM measurement. This work was financially supported by NSF (20773074, 21033004 & 20921001) of China.Notes and references
- M. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293 CrossRef CAS.
- E. Roduner, Chem. Soc. Rev., 2006, 35, 583 RSC.
- T. Kiyonaga, M. Fujii, T. Akita, H. Kobayashi and H. Tada, Phys. Chem. Chem. Phys., 2008, 10, 6553 RSC.
- R. Elghanian, J. J. Storhoff, R. C. Mucic, R. L. Letsinger and C. A. Mirkin, Science, 1997, 277, 1078 CrossRef CAS.
- S. Chen, R. S. Ingrma, M. J. Hostetler, J. J. Pietron, R. W. Murray, T. G. Schaaff, J. T. Khoury, M. M. Alvarez and R. L. Whetten, Science, 1998, 280, 2098 CrossRef CAS.
- H. Winnischofer, T. C. Rocha, W. C. Nunes, L. M. Socolovsky, M. Knobel and D. Zanchet, ACS Nano, 2008, 2, 1313 CrossRef CAS.
- G. C. Bond and D. T. Thompson, Catal. Rev. Sci. Eng., 1999, 41, 319 CrossRef CAS.
- F. Porta, L. Prati, M. Rossi, S. Coluccia and G. Martra, Catal. Today, 2000, 61, 165 CrossRef CAS.
- D. Zhao, Y. H. Wang and B. Q. Xu, J. Phys. Chem. C, 2009, 113, 20903 CrossRef CAS.
- M. Turner, V. B. Golovko, O. P. Vaughan, P. Abdulkin, A. Berenguer-Murcia, M. S. Tikhov, B. F. Johnson and R. M. Lambert, Nature, 2008, 454, 981 CrossRef CAS.
- N. Shalkevich, W. Escher, T. Bu, B. Michel, L. Si-Ahmed and D. Poulikakos, Langmuir, 2010, 26, 663 CrossRef CAS.
- W. Chen and S. W. Chen, Angew. Chem., Int. Ed., 2009, 48, 4386 CrossRef CAS.
- C. A. Witham, W. Y. Huang, C. K. Tsung, J. N. Kuhn, G. A. Somorjai and F. D. Toste, Nat. Chem., 2010, 2, 36 Search PubMed.
- R. Narayanan and M. A. El-Sayed, Top. Catal., 2008, 47, 15 CrossRef CAS.
- We have proposed to use Pt utilization (UPt) instead of Pt dispersion to raise the ever increasing concern on saving precious metal resources in catalysis, although both UPt and Pt dispersion refer to the same property (percentage exposed for a given Pt catalyst). The UPt sounds clearer to general readers about the usability of precious metal atoms for a special catalysis or in a chemical reaction. A thorough description of UPt and its measurement are provided in ref. 16 and 22.
- D. Zhao and B. Q. Xu, Angew. Chem., Int. Ed., 2006, 45, 4955 CrossRef CAS.
- Y. Li and M. A. El-Sayed, J. Phys. Chem. B, 2001, 105, 8938 CrossRef CAS.
- J. N. Kuhn, W. Huang, C. K. Tsung, Y. W. Zhang and G. A. Somorjai, J. Am. Chem. Soc., 2008, 130, 14026 CrossRef CAS.
- A. Villa, D. Wang, D. S. Su and L. Prati, ChemCatChem, 2009, 1, 510 Search PubMed.
- J. N. Kuhn, C. K. Tsung, W. Huang and G. A. Somorjai, J. Catal., 2009, 265, 209 CrossRef CAS.
- C. Mohr, H. Hofmeister and P. Claus, J. Catal., 2003, 213, 86 CrossRef.
- D. Zhao and B. Q. Xu, Phys. Chem. Chem. Phys., 2006, 8, 5106 RSC.
- Y. H. Wang, D. Zhao and B. Q. Xu, Chin. J. Catal., 2008, 29, 297 Search PubMed.
- H. Tsunoyama, H. Sakurai, N. Ichikuni, Y. Negishi and T. Tsukuda, Langmuir, 2004, 20, 11293 CrossRef CAS.
- X. W. Yang, G. R. Zhang, Y. X. Li and B. Q. Xu, Acta Phys. -Chim. Sin., 2009, 25, 2565 Search PubMed.
- D. Zhao, B. Yan and B. Q. Xu, Electrochem. Commun., 2008, 10, 884 CrossRef CAS.
- D. Zhao, Y. H. Wang, B. Yan and B. Q. Xu, J. Phys. Chem. C, 2009, 113, 1242 CrossRef CAS.
- H. Tsunoyama, N. Ichikuni, H. Sakurai and T. Tsukuda, J. Am. Chem. Soc., 2009, 131, 7086 CrossRef CAS.
- M. Okumura, Y. Kitagawa, T. Kawakami and M. Haruta, Chem. Phys. Lett., 2008, 459, 133 CrossRef CAS.
- H. H. Huang, X. P. Ni, G. L. Loy, C. H. Chew, K. L. Tan, F. C. Loh, J. F. Deng and G. Q. Xu, Langmuir, 1996, 12, 909 CrossRef CAS.
- Y. Borodko, S. E. Habas, M. Koebel, P. Yang, H. Frei and G. A. Somorjai, J. Phys. Chem. B, 2006, 110, 23052 CrossRef CAS.
- L. Qiu, F. Liu, L. Zhao, W. Yang and J. Yao, Langmuir, 2006, 22, 4480 CrossRef CAS.
- A. M. Van der Putten, J. G. de Bakker and L. G. Fokkink, J. Electrochem. Soc., 1992, 139, 3475 CrossRef CAS.
- C. Aliaga, J. Y. Park, Y. Yamada, H. S. Lee, C. Tsung, P. Yang and G. A. Somorjai, J. Phys. Chem. C, 2009, 113, 6150 CrossRef CAS.
- X. Ding, Z. Li, J. Yang, J. G. Hou and Q. Zhu, J. Chem. Phys., 2004, 120, 9594 CrossRef CAS.
- M. B. Torres, E. M. Fernandez and L. C. Balbas, J. Phys. Chem. A, 2008, 112, 6678 CrossRef CAS.
- V. Stamenkovic, B. S. Mun, K. J. Mayrhofer, P. N. Ross, N. M. Markovic, J. Rossmeisl, J. Greeley and J. K. Norskov, Angew. Chem., Int. Ed., 2006, 45, 2897 CrossRef CAS.
- I. Martin, T. Skalicky, J. Langer, H. Abdoul-Carime, G. Karwasz, E. Illenberger, M. Stano and S. Matejcik, Phys. Chem. Chem. Phys., 2005, 7, 2212 RSC.
- J. D. Lovic, A. V. Tripkovic, S. L. Gojkovic, K. D. Popovic, D. V. Tripkovic, P. Olszewski and A. Kowal, J. Electroanal. Chem., 2005, 581, 294 CrossRef CAS.
- H. Igarashi, T. Fujino, Y. Zhu, H. Uchida and M. Watanabe, Phys. Chem. Chem. Phys., 2001, 3, 306 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Preparation conditions of Au colloids (Table S1). Extended TEM characterization (Fig. S1). Complete cyclic voltammetry curves of formic acid electrooxidation (Fig. S2). See DOI: 10.1039/c0nr00295j |
| This journal is © The Royal Society of Chemistry 2010 |