Functionalized gold nanoparticles: a detailed in vivo multimodal microscopic brain distribution study†
Fernanda
Sousa‡
a,
Subhra
Mandal
b,
Chiara
Garrovo
a,
Alberto
Astolfo
c,
Alois
Bonifacio
d,
Diane
Latawiec§
e,
Ralf Hendrik
Menk
f,
Fulvia
Arfelli
cg,
Sabine
Huewel
h,
Giuseppe
Legname
e,
Hans-Joachim
Galla
h and
Silke
Krol‡
*a
aNanoBioMed Lab @ LANADA (Laboratory for Nanodiagnostics, Drug Delivery and Analysis), CBM - Cluster in Biomedicine S.c.r.l., Basovizza, AREA Science Park, Trieste, Italy. E-mail: silke.krol@istituto-besta.it
bSISSA - Scuola Internazionale Superiore di Studi Avanzati -International School of Advanced Studies, Trieste, Italy
cDepartment of Physics, University of Trieste, Trieste, Italy
dCentre of Excellence for Nanostructured Materials, University of Trieste, Trieste, Italy
eLaboratory of Prion Biology, Neurobiology Sector, Scuola Internazionale Superiore di Studi Avanzati - International School of Advanced Studies, AREA Science Park, Trieste, Italy
fSincrotrone Trieste S.C.p.A. di Interesse Nazionale, AREA Science Park, Trieste, Italy
gINFN-Istituto Nationale di Fisica Nucleare, Trieste, Italy
hInstitute for Biochemistry, Westf, Wilhelms-University, Muenster, Germany
First published on 15th October 2010
Abstract
In the present study, the in vivo distribution of polyelectrolyte multilayer coated gold nanoparticles is shown, starting from the living animal down to cellular level. The coating was designed with functional moieties to serve as a potential nano drug for prion disease. With near infrared time-domain imaging we followed the biodistribution in mice up to 7 days after intravenous injection of the nanoparticles. The peak concentration in the head of mice was detected between 19 and 24 h. The precise particle distribution in the brain was studied ex vivo by X-ray microtomography, confocal laser and fluorescence microscopy. We found that the particles mainly accumulate in the hippocampus, thalamus, hypothalamus, and the cerebral cortex.
Introduction
A large number of compounds of pharmaceutical interest are tested nowadays for the treatment of neurodegenerative disorders. However, most of these compounds which demonstrate efficacy in vitro are not able to reach the central nervous system (CNS), at least not in pharmacologically significant concentrations.1 Delivery of therapeutic agents to the CNS is hindered by the blood–brain barrier (BBB), which is composed of a tightly sealed layer of endothelial cells and astrocytes, regulating the permeation and diffusion of drugs from the blood stream into the brain.1,2Different approaches to deliver to the CNS compounds that do not normally cross the BBB have been attempted by various research groups. One approach is the temporary opening of the tight junctions using a hyperosmolaric solution.3 However, this technique carries serious adverse effects associated with the frequency of administration of these hyperosmolar agents. Other approaches involve the direct delivery of therapeutics into the brain by intracerebral injection into the cerebral parenchyma or cerebral ventricles. Notwithstanding that these are invasive approaches associated with adverse tissue reaction and haemorrhage, the amount of therapeutic agents that can be administered at one time is very limited and may also be subject to limited diffusion of the agents away from the injection site.2–4
The barrier function of the BBB is not absolute, as the brain needs nutrients, oxygen and essential molecules. Complex and highly regulated, the BBB screens the biochemical, physicochemical and structural features of solutes in its periphery, thus affording barrier selectivity in the passage of molecules into the brain parenchyma. Their passage may be mediated by simple diffusion or specific transport (adsorptive, receptor- or carrier-mediated transport mechanisms), both of which can be affected by interactions in the blood compartment (outside the BBB) and within the endothelial cells (at the BBB level).5–8
With the growing knowledge about the nature of these mechanisms, several current delivery and targeting strategies are designed to use existing pathways. They include the development of hydrophobic or receptor-mediated prodrugs (drug molecules covalently bound either to a hydrophobic linker or to glucose molecules) which are cleaved in the brain, or liposomal drug preparation and even targeting moieties can be bound, such as antibodies.9–12 However, despite the fact that some of these approaches have achieved increased drug delivery into the brain, the therapeutic efficacy decreases either due to chemical modifications or a low drug load, thus limiting their usefulness.
Polyelectrolyte multilayer coated nanoparticles are promising candidates as drug carriers, thanks to their ability to adsorb and incorporate drug molecules, resulting in a high local drug concentration sufficient to treat a small number of cells in the direct vicinity of the particles, and the possibility of engineering the surface functionalities for specific targeting. Gold nanoparticles were chosen here as the core due to the fact that they are non-toxic in the selected size range, monodisperse and easy to functionalize through polyelectrolyte multilayer encapsulation.13–15 Moreover, the luminescence of gold and its electron density allow its visualization and characterization by UV-vis spectrophotometry, two-photon excitation fluorescence microscopy, X-ray computer microtomography (CT), and transmission electron microscopy (TEM).
Previous experiments which reported the entry of citrate-stabilized gold nanoparticles (AuNPs) into the brain after intravenous administration16,17 showed that only gold nanoparticles with a diameter of 15 nm or 50 nm could pass through the BBB. However, little is known about the distribution of the particles in the brain.
We previously developed polyelectrolyte multilayer-coated gold nanoparticles which can potentially be used for the treatment of prion disease as they exhibit two therapeutically relevant functional groups, sulfonates and primary amines,18 in varying ratios on the surface. In vitro, however, these polyelectrolytes proved to be cytotoxic to a confluent layer of primary porcine brain capillary endothelial cells (PBCEC).13 In the present work we modify the coating in order to protect the endothelial cells by incorporating human serum albumin (HSA, a natural blood component; >60% of plasma protein; 400 to 675 μM) and test the effect on an artificial model for the blood–brain barrier. We expect an additional beneficial effect of the HSA coating in minimizing recognition by the immune system and increasing the circulation time of the coated NPs. Furthermore, it has been shown that a cationized form of HSA can induce absorptive-mediated transcytosis through the BBB.11,12,19
The coated gold nanoparticles were injected into the tail vein of healthy mice and the biodistribution was characterized in detail. The whole body distribution was determined by NIR-TD (near infrared time-domain) measurements in living animals. Ex vivo X-ray microtomography of the whole brain allowed the identification of areas with high AuNP concentration whilst confocal laser scanning microscopy (CLSM), optical fluorescence and light microscopy of brain slices verified that the nanoparticles were localized to specific brain regions.
Results and discussion
Characterization of nanoparticles
The biodistribution of nanoparticles in vivo is largely influenced by the physicochemical characteristics, e.g. particle size and surface properties (i.e. charge).15,16,20 Thus, characterization of nanoparticle properties and the analysis of its distribution constitute two very important steps in determining their potential as therapeutic/drug carriers. Monodisperse gold nanoparticles of diameter 15 ± 1 nm were prepared according to the technique of Turkevich.21 The citrate-stabilized AuNP was coated by electrostatic driven self-assembly of oppositely charged polyions (polycation: PAH, polyallylamine hydrochloride, 15 kDa; polyanion: PSS, polystyrenesulfonate, 4.3 kDa) or the negative net charge of HSA (3 domains: 2 negatively charged, 1 positively charged). The detailed coating procedure has been previously described by Chanana et al.,13 and Schneider and Decher.14 The AuNPs coated with HSA and polyelectrolytes are referred to as PNP in the following while those without protein are referred to as 2A because they consist of 2 polyelectrolyte layers with the last one being PAH. The physical parameters of NPs used for the in vivo and in vitro studies are provided in Table 1.| D h /nm | ζ-potential/mV (in water at pH 7.4) | Conc.b/nM | Number of particles per ml | |
|---|---|---|---|---|
| Mean ± SD | Mean ± SD | |||
| a D h hydrodynamic diameter as determined by dynamic light scattering. b The particle concentration was calculated by UV-vis absorption applying the Beer–Lambert law (λabs at 518 nm and ε = 5.14 × 107 M−1 cm−1).22 | ||||
| PNP (cytotoxicity/injected) | 115 ± 5 | +21.5 ± 4.5 | 144.5 ± 43 | 8.7 ± 2.6 × 1013 |
| 2A (cytotoxicity) | 126.5 ± 6 | +60.5 ± 2 | 46 ± 7 | 2.76 ± 0.42 × 1013 |
For each coating step dynamic light scattering (DLS) and ζ-potential measurements were performed in order to determine the hydrodynamic diameter (Dh), polydispersity index (PDI), and the surface charge. The change in charge and size after adding each layer confirmed the deposition of the oppositely charged polyelectrolytes.13 The relatively lower positive surface charge for the NPs with HSA and PAH co-adsorbed indicated the presence of the protein on the surface of the nanogold (Table 1).
Using surface-enhanced Raman scattering (SERS), the binding of albumin was confirmed by detection of the fluorescein isothiocyanate (FITC) signal of FITC covalently bound to HSA (Fig. 1a). The SERS spectrum of PSS/PAH + FITC-HSA coated AuNPs and that of gold NPs coated only with PSS/PAH (Fig. 1b) are remarkably different. Moreover, the spectrum of the NPs incubated with the FITC-labeled HSA presents several intense bands with frequencies very similar to those observed in the Raman spectrum of a FITC solution (Fig. 1c). Differences between the SERS spectrum of FITC-labeled HSA on gold nanoparticles and the Raman spectrum of FITC in solution, in the relative intensity of bands, are likely to be due to surface selection rules due to the presence of the metal surface23 and from the modification of the structure of FITC upon protein conjugation.
 | ||
| Fig. 1 SERS spectra (900–1800 cm−1 region) of coated gold nanoparticles. Gold nanoparticles coated (a) with PSS/PAH + FITC-labeled HSA, and (b) with PSS/PAH, together with the result of their spectral subtraction (a) − (b). A pre-resonant Raman spectrum of a 0.1 mg ml−1 FITC aqueous solution (c) is shown for comparison. | ||
The DLS, ζ-potential and SERS data clearly imply the presence of albumin on the surface of coated AuNPs.
Influence of human serum albumin coating on nanoparticle toxicity - transendothelial electrical resistance
The major factors influencing the potential toxicity of nanoparticles are their size, charge, chemical composition, surface chemistry/functionalization and aggregation tendency.24,25 A well-established technique to study the toxicity of particles, or drugs, to the vessels of the blood–brain barrier in vitro is measuring the transendothelial electrical resistance (TEER). TEER measurements have been previously performed on a primary culture of a confluent layer of PBCEC (porcine brain capillary endothelial cells) as an in vitro model for the BBB.26,27 Accordingly, we used TEER measurements to compare the toxicity of the novel HSA coated nanoparticles to that of polyelectrolyte coated ones.Nanoparticles were added to the medium in the apical compartment to give final concentrations of 0.08 nM, 1.28 nM, and 7.2 nM. The nanoparticles were injected into mice to a final concentration of 7.2 nM assuming the mouse blood volume to be 5 mL. The control was a confluent layer of PBCEC which received the same volume of medium as injected into the treated cells. The changes observed in the TEER on the endothelial cell layer clearly indicate that the toxicity of the particles is concentration-dependent (Fig. 2). In general it can be stated that 2A nanoparticles (without HSA) (Fig. 2b) showed, at concentrations of 1.28 nM and 7.2 nM, higher toxicity as indicated by the faster response of the cell layer to the particles when compared to the polyelectrolyte/protein coated nanoparticles (PNP) (Fig. 2a). After the addition of nanoparticles at a concentration of 7.2 nM, the resistance almost immediately decreases to zero, whereas a moderate concentration of 1.28 nM leads to a delayed and slower decrease of the resistance over time. In contrast, the 0.08 nM concentration of coated AuNPs gives rise to a similar or in some experiments even a higher resistance of the PBCEC layer than the control cells.
 | ||
| Fig. 2 The cytotoxic effect of the nanoparticles on a primary PBCEC culture, as revealed by the decrease in TEER measurement, is dependent on the concentration. a) Polyelectrolyte/protein coated nanoparticles (PNP) and b) nanoparticles coated with PSS/PAH (2A). TEER measurements were performed in a medium supplemented with nanoparticles. All values were normalized to the control (medium without nanoparticles). Data are given as means ± SD (n = 6). | ||
Whilst the toxicity appears to be dependent on the particle concentration, we do not expect a significant toxic effect in the animal because some particles partially distribute in other organs, not all the particles reach the BBB at the same time and only the highest concentration showed a dramatic toxicity. This was supported by the fact that after injection and over the complete experimental duration of 7 days for the long-term experiments we did not observe any alteration in the behaviour of the animals indicating toxicity or barrier damage. The precise particle concentration which arrives in the brain microvessels is not yet known but experiments to determine it are underway.
Biodistribution of nanoparticles
Fluorescence could be detected in the mouse head within 30 min (Fig. 3) after particle administration. However, the differences in intensity between 0 h and 48 h are very small and therefore it was observed that the temporal appearance of a peak concentration was not very reproducible. This was followed by a decrease in the signal to nearly normal background intensity after 7 days (Fig. S1†). The continuous high signal for up to 48 h confirms a long circulation time of the nanoparticles, probably induced by the albumin preventing recognition by the immune system. The signal decrease after 24 h could be explained either by enzymatic degradation of albumin to which the fluorophore is covalently bound or by particles being released from the brain. A detailed study into the uptake kinetics and a more detailed time course will be addressed in the future. Due to the fact that for a very short time we assume that the particles have not yet entered the brain, in the following we sacrificed the mice after 19 h to allow passage and adequate distribution in the brain.
 | ||
| Fig. 3 In vivo PNP fluorescence intensity of a mouse head up to the maximum signal intensity time of 19 h. The intensity of the untreated control was compared to that at 30 min and 19 h after particle injection. The intensity is shown after background correction of the intrinsic tissue autofluorescence of the control mouse. | ||
One of the drawbacks of NIR-TD imaging is that it has a spatial resolution limit of about 0.5 mm. Whilst a clear accumulation of nanoparticles in the head can be seen we cannot determine outright whether the particles have crossed the BBB and where precisely they are located in the brain. In order to localize the PNPs more precisely in the brain, we carried out further analysis on the ex vivo brains using X-ray microtomography, confocal laser scanning microscopy (CLSM), and epifluorescence microscopy. To ascertain whether the particles observed in NIR-TD are the same as those visualized in CLSM we double labeled the PNPs. Cy5.5 was covalently bound to HSA and FITC linked to PAH for fluorescence microscopy whilst the X-ray signal came directly from the gold core itself. Mainly we focused on samples from animals (n = 13) sacrificed 19 h after injection.
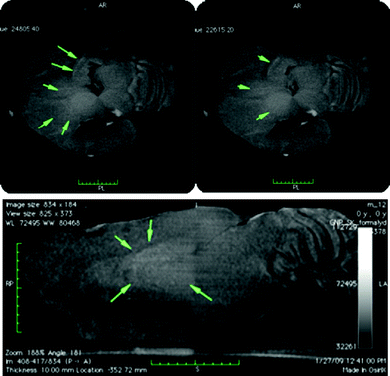 | ||
| Fig. 4 Tomographic reconstructions of a mouse brain removed 19 h after injection of PNPs. The upper panel shows the transverse plane and the lower panel depicts the sagittal plane. The two transverse planes in the upper panel are separated by 200 μm. Green arrows highlight the thalamus and hypothalamus, areas in which in the axial and sagittal planes a higher contrast and subsequently higher nanogold concentration was found. | ||
While the untreated mouse does not show any significant contrast (Fig. S2†), mice sacrificed 19 h after PNP injection revealed an area of contrast comprising the entire region of the thalamus/hypothalamus (green arrows in Fig. 4), indicating an accumulation of AuNPs. In addition a thin line of higher contrast at the boundary of the thalamus was visible. The mice sacrificed 7 days after nanoparticle injection revealed no contrast in the thalamus/hypothalamus complex. However, two thin lines of higher absorption/contrast were prominent indicating higher gold concentrations on the boundary of lamellae separating the different thalamic subparts (green arrows in Fig. S3†).
 | ||
| Fig. 5 CLSM images of an entire brain slice in (a) low (20× magnification) resolution. The cerebral cortex was imaged in (b) higher magnification (square). The presence of PNPs was proven by a spectrometric analysis (e) of the highly fluorescent clusters marked in (c) and the presence of the FITC signal (d). The cellular distribution of the nanoparticles in specific neuronal regions of the brain was visualized using a combination of different labels (f and g). The green bright dots are the PNPs labeled with FITC (arrows); cell bodies of glia cells and neurons (Nissl) are labeled red and the nucleus (DAPI) blue. The general green color of the tissue is the autofluorescence of the cells. The left images (f1 and g1) depict the double stain Nissl/FITC while the right images (f2 and g2) show DAPI/FITC. Images f1 and f2 are from the cortex, and g1 and g2 are Purkinje cells which accumulate particles (arrows) while the cerebellum only shows autofluorescence. | ||
The localization and cellular distribution pattern of nanoparticles in sectioned brain tissue was visualized by epifluorescence microscopy using a combination of three different markers: Nissl, DAPI, and FITC. DAPI is used to selectively label the nuclei whereas Nissl stains the cell body of neurons and glia. The nanoparticles were visualized from the FITC signal. The double-stained brain slices were visualized with a combination of bright field white light and epifluorescence microscopy using different emission filters to visualize the various markers.
Some merged fluorescent micrographs of selected areas of the brain which revealed a strong/bright FITC signal, i.e. accumulation of PNPs in the form of clusters probably in vesicular cellular structures (bright green punctuate appearance) of the brain slice, co-stained with Nissl (red) or with DAPI (blue) are depicted in Fig. 5f of the cortex and Fig. 5g of the cerebellum. It has to be mentioned that homogeneously distributed single nanoparticles in the cytosol will not be identifiable in optical microscopy but will contribute to the background signal. In particular, regions of the brain that had revealed high nanoparticle concentrations by X-ray and CLSM were analyzed in more detail to assess subcellular nanogold distribution. The presence and co-localization of both the FITC signal (PNP) and the neurons or glia identified by the Nissl stain (i.e. red) clearly proved that the particles cross the blood–brain barrier and enter the cells by endocytosis (clustering of the NPs). From the second double-staining with DAPI and FITC it can be concluded that the particle do not enter the nucleus.
The regions where the particles were visualized at a cellular level were essentially the same as those indicated by CLSM and X-ray tomography. Most interesting for the treatment of prion disease but also Alzheimer's disease is the cerebral cortex.27 A high concentration of particles can be observed in the cortex itself (Fig. 5f) and in the Purkinje cells (Fig. 5g, arrows). Purkinje cells are a class of GABAergic neurons located in the cerebral cortex close to the cerebellum. In contrast, no significant clustering of nanoparticles can be detected within the cerebellum, which could be due to the detection limit of the microscope (only clusters bigger than 300 nm are visible) or the absence of particles (Fig. 5g).
Other regions of the brain also showed accumulation of PNPs. In the hippocampus, there was an accumulation of the nanoparticles in all cellular layers: the pyramidal cell layer of the CA1 (Fig. S5a†) and CA3 regions (Fig. S5b†), as well as in the granule cell layer of the dentate gyrus (Fig. S5c†). Particles were found in the thalamus (Fig. S5d†) and, interestingly, in the medulla, towards the spinal cord (Fig. S5e†).
A detailed distribution of gold nanoparticles able to cross the blood–brain barrier and accumulate in specific highly defined areas of the brain has been shown here for the first time to our knowledge. In our study we followed the biodistribution of nanogold particles in mice, starting at a whole body level down to the subcellular distribution in specific cells, using complementary imaging methods. Moreover, we showed that PSS/PAH + HSA coated nanogold particles do pass through the blood–brain barrier and distribute heterogeneously throughout the brain, indicating an active uptake rather than a barrier damage induced leakage. In particular, high particle concentrations were detected in the hippocampus, hypothalamus and thalamus regions, but also in the cortex. The localization of the potentially drug-like nanogold particles in the cortex and the hypothalamus are of special interest as they are close to the areas in which Alzheimer's (cerebral cortex),30 Parkinson's (substantia nigra),31 and prion disease originate. In these regions in particular, we deduce from the fluorescence microscopy images that specific cells are taking up the particles, most probably by endocytosis, but that the particles are not entering the nucleus. More detailed studies of the particular mode of entry into the cells are planned for future work.
Experimental
Synthesis of gold nanoparticles
Monodisperse gold nanoparticles were prepared following the technique of Turkevich.21 In brief, for nanogold of 15 ± 1 nm diameter, 5.3 mg of NaAuCl4·2H2O in 25 ml of Milli-Q grade water was boiled under reflux, before a rapid addition of 1 ml of a 1% citrate solution. The solution was then kept boiling for 20 min. After cooling down to room temperature the solution was stored, protected from light. The particle concentration was calculated by UV-vis absorption applying the Beer–Lambert law (λabs at 518 nm and ε = 5.14 × 107 M−1 cm−1 found by Liu et al.22) All experiments described in this manuscript were performed with this gold nanoparticle stock solution (31.8 nM).Polyelectrolyte coating
The polyelectrolyte coating was applied in accordance with the method previously described by Schneider and Decher14 and Chanana et al.13 with only a few modifications. The polyelectrolytes PAH (polyallylamine hydrochloride, 15 kDa), PAH-FITC and PSS (polystyrene-4-sulfonate, sodium salt, 4.3 kDa) were used for the coating procedure. Briefly, for the first layer 1 mL of colloidal nanogold was added drop-wise under constant stirring to 200 μL of PSS solution (10 mg mL−1). After incubation for 20 min in the dark, this solution was centrifuged for 20 min at 20,000 × g. The supernatant was removed and the coated particles, a deep red gel-like pellet, were washed twice by centrifugation/resuspension in Milli-Q water. Prior to the next layer deposition the coated nanogold was stored in the dark for 1 h. For particles labeled with 2A, the PSS-coated nanogold particles were added drop-wise under constant vortexing to 500 μL of PAH solution (3 mg mL−1) followed by washing steps.For the injection into mice the particles (PNP) needed an additional layer of human serum albumin (HSA) to enable the blood–brain barrier cross-over. The second layer was self-assembled by a co-deposition of PAH with HSA at pH 7.4.
For the co-absorption and visualization in NIR-TD imaging and fluorescence microscopy, 500 μL of FITC-PAH (1 mg mL−1) and 500 μL of HSA (1 mg mL−1) covalently labeled with Cy5.5, a dye which can be visualized in the eXplore Optix pre-clinical imager with λex = 670 nm and λem = 700 nm, were mixed drop-wise. A volume of 500 μL of the mixture was added drop-wise and under continuous vortexing to PSS-coated nanogold. Each coating step was monitored by dynamic light scattering (DLS) and ζ-potential measurements on a ZS Zetasizer (Malvern, Milan, Italy) to determine the hydrodynamic diameter (Dh), polydispersity index (PDI) and surface charge. Finally, the coated nanoparticles were concentrated from 4 mL to 500 μL, prior to the washing steps for 40 min at 10,000 × g.
Surface-enhanced Raman scattering
Surface-enhanced Raman scattering (SERS) and pre-resonant Raman spectra were collected using an InVia Raman system (Renishaw plc, Wotton-under-Edge, UK). A laser (632.8 nm He–Ne laser, Melles-Griot, Albuquerque NM, USA) was focused by a 10× microscope objective (0.25 NA) on the sample, consisting of a 50 μL drop of the gold nanoparticle dispersion deposited on a CaF2 slide (OEC Optoelectronic Components GmbH, Zusmarshausen, Germany) for SERS measurements or aqueous solutions of FITC (0.1 mg mL−1) for pre-resonant Raman. The Raman scattered light was then collected by InVia microscope optics, analyzed and detected with the built-in spectrograph equipped with a 1800 g mm−1 holographic grating, yielding a spectral resolution of 4 cm−1. The laser power applied to the sample was 15 mW. The total acquisition time was 60 s per spectrum.Preparation and cultivation of primary porcine brain capillary endothelial cells
Porcine brain capillary endothelial cells (PBCEC) were isolated, cultured and cryo-conserved as described previously.29 In brief, the cerebra of freshly slaughtered adult pigs were separated from the meninges and mechanically homogenized. The brain homogenate was digested with 6.5% (v/v)-protease/dispase II (from B. polymyxa, Becton Dickinson, Heidelberg, Germany) in preparation medium (Medium 199 Earle supplemented with 0.7 mM L-glutamine, 100 μg mL−1 gentamicin, 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin, all from Biochrom, Berlin, Germany). Furthermore, dextran density centrifugation was applied to separate the brain capillaries from myelin and cell debris. The resuspended pellet was filtered through a nylon mesh (180 μm pore size) after trituration of the capillary. The basement membrane was removed by another enzymatic digestion with 1% (w/v) collagenase/dispase II in plating medium (preparation medium supplemented with 10% (v/v) newborn calf serum, NCS, Biochrom, Berlin, Germany). The endothelial cells were further purified using a discontinuous Percoll density gradient and were then seeded in plating medium containing puromycin (2.5 μg mL−1, Sigma-Aldrich, Munich, Germany) on collagen G-coated culture flasks (Nunc, Roskilde, Denmark and Wiesbaden, Germany). Twenty four hours after initial plating, cells were washed with phosphate buffered saline (PBS) containing 1 mM Ca2+ and 0.5 mM Mg2+ and supplied with fresh plating medium without gentamicin but with puromycin (2.5 μg mL−1, Sigma-Aldrich, Munich, Germany) to remove pericytes. Primary cultures of PBCEC were subcultured by gentle trypsinization (0.063% w/v) at room temperature on day 2 in vitro (DIV 2) in order to reduce contamination by other cells. Afterwards PBCEC were frozen and stored in liquid nitrogen. For experiments the cells were gently thawed, suspended in plating medium (Medium 199 Earle containing 0.7 mM L-glutamine, 1(2)00 U mL−1 penicillin, 100 μg mL−1 streptomycin, 100 μg mL−1 gentamycin and 10% (v/v) newborn calf serum, all Biochrom, Berlin, Germany) and seeded on filters (Costar® Transwell™; Corning, Schiphol, The Netherlands and Wiesbaden, Germany; 0.4 μm pores; 1.13 cm2 growth area) pre-coated with rat tail collagen. After 48 h the cell layers reached confluence and the plating medium was replaced by chemically defined medium (DMEM/Ham's F 12 containing 4.1 mM L-glutamine, 100 U mL−1 penicillin, 100 μg ml−1 streptomycin, 100 mg mL−1 gentamycin, all Biochrom, Berlin, Germany, and with 550 nM hydrocortisone, Sigma-Aldrich, Taufkirchen, Germany).Transendothelial electrical resistance measurements
For transendothelial electrical resistance (TEER) determination filter inserts with nanoporous polycarbonate membranes (Costar® Transwell™; Corning, Schiphol, The Netherlands; 0.4 μm pores; 1.13 cm2 growth area) were coated with rat-tail collagen and PBCEC were seeded in a density of 250,000 cells cm−2 and cultured as described above. For TEER measurements over time a device reading 24 electrodes in parallel (CellZscope®, NanoAnalytics, Münster, Germany) was used which allows automated long-term monitoring and the application of nanoparticles during the measurement.In vivo biodistribution of coated particles in a healthy mouse
The experiments were performed on male CD1 mice of approximately 30 g and 6 weeks of age. The animals were obtained from Harlan Laboratories (Udine, Italy) and maintained under pathogen-free conditions. All the experimental procedures were performed according to the guidelines of the European (86/609/EEC) and the Italian (D.L.116/92 and subsequent addenda) laws and approved by the Italian Ministry of University and Research.The day before the treatment, the mice were anesthetized by an intramuscular injection of a diluted mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 in PBS) composed of 0.4 mL Zoletil 100 and 0.25 mL Rompun 2% (3 μL g−1 body weight), and shaved in the regions of interest to avoid laser light scattering caused by hair. The following day, the mice were anesthetized using a gaseous anesthesia system (2Biological Instruments, Italy), based on isoflurane mixed with oxygen and nitrogen protoxide. Anesthesia was first induced with 2% isoflurane in a pre-anesthesia chamber and then the mouse was placed inside the eXplore Optix while the anesthesia was maintained with 1% isoflurane.
:5 in PBS) composed of 0.4 mL Zoletil 100 and 0.25 mL Rompun 2% (3 μL g−1 body weight), and shaved in the regions of interest to avoid laser light scattering caused by hair. The following day, the mice were anesthetized using a gaseous anesthesia system (2Biological Instruments, Italy), based on isoflurane mixed with oxygen and nitrogen protoxide. Anesthesia was first induced with 2% isoflurane in a pre-anesthesia chamber and then the mouse was placed inside the eXplore Optix while the anesthesia was maintained with 1% isoflurane.
The mice were then injected with 150–200 μL nanoparticles into the vein in the tail. At specific time intervals, the mice were scanned with the eXplore Optix in order to observe the distribution pattern of the nanoparticles throughout the body and more specifically the brain uptake. Controls usually received no particles but in one case Cy5.5-labelled albumin was injected.
In vivo near infrared time-domain optical imaging
In vivo time-domain fluorescence measurements were performed with an eXplore Optix pre-clinical imager with a 670 nm pulsed laser diode, a repetition frequency of 80 MHz and a time resolution of 12 ps light pulse for excitation. Fluorescence emission was collected at 700 nm and detected through a fast photomultiplier tube and a highly sensitive time-correlated single-photon counting system. Two-dimensional scanning regions of interest (ROI) were selected, and the laser power, integration time and scan step were optimized according to the emitted signal. The data were recorded as temporal point-spread functions, and the images were reconstructed as fluorescence intensity and fluorescence lifetime. Prior to injecting the probe, the mice were imaged to obtain the background signal. With this experimental set-up we were able to follow the distribution of the particles for 7 days after injection.Tissue preparation
The mice were sacrificed by cervical dislocation, the brain was removed from the skull, and washed in PBS. For confocal microscopy, the brains were placed in 1% zinc fixative for 6 h and then transferred to a solution containing 1% zinc fixative and 30% sucrose overnight. The brains were then embedded into Tissue-Tek® O.C.T.™ Compound (Sakura Finetek), a convenient specimen matrix for cryostat sectioning, and stored at −80 °C until microtome slicing. The brains were sectioned in a sagittal orientation with a microtome (Bio-Optica, Germany) in 10 μm sections and thaw-mounted onto surface-treated glass slides.A number of sections were prepared for Nissl and DAPI staining. Nissl staining labels the cell bodies of neurons and glia cells brilliant violet which when visualized in fluorescence appears red. Nissl staining was performed following the standard protocol. Sections were rehydrated in xylene (2 times for 3 min), followed by decreasing concentrations of ethanol (100%, 95%, 75%, 3 min each) before washing briefly with Milli-Q grade water for 30 s. They were then immersed in 0.25% cresyl violet for 10 min. The excessive staining was washed with Milli-Q water and sections were dehydrated in solutions with increasing concentration of ethanol (EtOH) (75%, 95% and 100% EtOH) and given a final wash for 45 min in xylene. After staining, the slides were coverslipped with a vector shield (Vector laboratories) and left to air dry at room temperature.
Staining with DAPI (4′-6-diamidino-2-phenylindole) was performed by adding a DAPI containing vector shield (Vector laboratories), directly to the mounted brain slices. After staining, the samples were examined with an epifluorescence microscope (Leica DM 6000B, Germany) using the appropriate filter cubes.
For X-ray imaging, the brains were fixed in 4% paraformaldehyde for 19 h followed by a series of sequential dipping through increasing concentrations of ethanol and finally with xylene (70% EtOH: 2 × 15 min, 90% EtOH: 2 × 15 min, 95% EtOH: 2 × 15 min, 100% EtOH: 2 × 30 min and xylene: 1 × 50 min) before embedding in paraffin.
Confocal laser microscopy
Imaging acquisition of the brain sections was performed with a Nikon C1 laser scanning confocal unit (Nikon D-eclipse C1Si, Japan) attached to an inversed fluorescence microscope with 100×/1.49 oil Apo TIRF objective (Nikon, Japan). Excitation was performed with an air-cooled Argon laser emitting at 488 nm and appropriate filters were used to collect the fluorescence emission. Images were acquired and processed using the operation software EZ-C1 for Nikon C1 confocal microscope.X-ray tomography
Mice brains were imaged utilizing the set-up (Fig. S6†) available in the recently installed TOMOLAB facility at the Sincrotrone Trieste based on a micro focus X-ray tube (Hamamatsu, 130 kV Microfocus X-ray Source, L6622-01, focal spot of about 5 μm). The imaging detector was a phosphor fiber optic coupled CCD (Photonic Science Ltd, X-ray Imager-VHR 60 camera system) with 10-micron pixels and an effective spatial resolution of 18 microns full width at half maximum of the point spread function. The pixel depth was 12 bits. For each projection, four images with an exposure time of 2.5 s each were averaged, resulting in an overall exposure time per projection of 10 s.The X-ray tube accelerating voltage was set to 50 keV and the current was held at 133 μA. A 0.5 mm Al filter was used to cut the low energy part of the spectrum resulting in a mean X-ray energy of about 24 keV. The source to object distance was set to 200 mm, whilst the source to detector distance was 450 mm yielding moderate phase contrast in the edge enhancement regime. The field of view was governed by the active area of the detector, which in this case was 52 mm by 48 mm. 900 projections were acquired equi-angularly over 360° resulting in an entrance dose in air of about 3 Gy. The dark and flat field corrected data were reconstructed using the COBRA software code from EXXIM (http://www.exxim-cc.com/Index.htm). Slice data were analyzed, manipulated and rendered using the Osirix software package (http://www.osirix-viewer.com) and ImageJ (http://rsb.info.nih.gov/ij/).
Conclusions
For the first time it was possible to show in detail that coated gold nanoparticles were able to cross the blood–brain barrier and enter defined neuronal structures. The areas in which high accumulation of particles was observed are close to areas in which protein (prion, beta-amyloid) aggregation and hence neurodegeneration takes place. Due to their multilayer functionalization the particles could be equipped with a small amount of drugs, RNA or targeting moieties. Moreover the visibility of gold by different imaging techniques also makes the system attractive for diagnostic purposes in the detection of brain-related diseases.Acknowledgements
This work was supported by a CIPE grant from the Italian government to F. S. and S. K., by Fondazione CRTrieste for the optical imaging facility (C. G.) and by a SISSA PhD fellowship for S. M. The authors wish to acknowledge Dr V. Lorusso for his valuable insights into nanoparticle distribution and the animal house staff of the University of Trieste. We would also like to acknowledge Dr F. Battisti for her support in tissue preparation. Moreover the authors are grateful to A. Balsamo and L. Moccagatta from NIKON for their generous help with the acquisition of the fluorescence spectra. A final thanks goes to G. Furlan for English editing of the manuscript.References
- W. M. Pardridge, Drug Discovery Today, 2007, 12, 54 CrossRef CAS.
- J. M. Koziara, P. R. Lockman, D. D. Allen and R. J. Mumper, J. Nanosci. Nanotechnol., 2006, 6, 2712 CrossRef CAS.
- E. A. Neuwelt and P. A. Barnet in Blood–brain barrier disruption in the treatment of brain tumors. Animal studies, ed. E. A. Neuwelt, Plenum, New York, 1989, pp 195–217 Search PubMed.
- W. Pan and A. J. Kastin, Brain Res. Rev., 2004, 46, 32 CrossRef CAS.
- J. Kreuter, Int. Congr. Ser., 2005, 1277, 85 CrossRef CAS.
- W. M. Pardridge, Adv. Drug Delivery Rev., 1999, 36, 299 CrossRef CAS.
- W. M. Pardridge, Bioconjugate Chem., 2008, 19, 1327 CrossRef CAS.
- U. Bickel, T. Yoshikawa and W. M. Pardridge, Adv. Drug Delivery Rev., 2001, 46, 247 CrossRef CAS.
- B. J. Spencer and I. M. Verma, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7594 CrossRef CAS.
- A. Zensi, D. Begley, C. Pontikis, C. Legros, L. Mihoreanu and S. Wagner, et al. , J. Controlled Release, 2009, 137, 78 CrossRef CAS.
- F. Xu, W. Lu, H. Wu, L. Fan, X. Gao and X. Jiang, J. Drug Targeting, 2009, 17, 423 Search PubMed.
- W. Lu, J. Wan, Z. She and X. Jiang, J. Controlled Release, 2007, 118, 38 CrossRef CAS.
- M. Chanana, A. Gliozzi, A. Diaspro, I. Chodnevskaja, S. Huewel and V. Moskalenko, et al. , Nano Lett., 2005, 5, 2605 CrossRef CAS.
- G. Schneider and G. Decher, Nano Lett., 2004, 4, 1833 CrossRef CAS.
- H. Labouta and M. Schneider, Int. J. Pharm., 2010, 395, 236 CrossRef CAS.
- W. H. De Jong, W. I. Hagens, P. Krystek, M. C. Burger, A. J. A. M. Sips and R. E Geertsma, Biomaterials, 2008, 29, 1912 CrossRef CAS.
- G. Sonavane, K. Tomoda and K. Makino, Colloids Surf., B, 2008, 66, 274 CrossRef CAS.
- C. R. Trevitt and J. Collinge, Brain, 2006, 129, 2241 CrossRef.
- A. K. Kumagai, J. B. Eisenberg and W. M. Pardridge, J. Biol. Chem., 1987, 262, 15214 CAS.
- B. D. Chithrani, A. A. Ghazani and W. C. W. Chan, Nano Lett., 2006, 6, 662 CrossRef CAS.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55 RSC.
- X. Liu, M. Atwater, J. Wang and Q. Huo, Colloids Surf., B, 2007, 58, 3 CrossRef CAS.
- X. X. Han, L. J. Cai, J. Guo, C. X. Wang, W. D. Ruan and W. Y. Han, et al. , Anal. Chem., 2008, 80, 3020 CrossRef CAS.
- N. Lewinski, V. Colvin and R. Drezek, Small, 2008, 4, 26 CrossRef CAS.
- J. H. Yen, H. S. Hsu and L. C. Tsai, Small, 2009, 5, 1553 CrossRef CAS.
- M. Erben, S. Decker, H. Franke and H.-J. Galla, J. Biochem. Biophys. Methods, 1995, 30, 227 CrossRef CAS.
- J. Wegener, M. Sieber and H.-J. Galla, J. Biochem. Biophys. Methods, 1996, 32, 151 CrossRef CAS.
- A. Abulrob, E. Brunette, J. Slinn, E. Baumann and D. Stanimirovic, Mol. Imaging, 2008, 7, 248.
- H. Franke, H.-J. Galla and C. T. Beuckmann, Brain Res. Protoc., 2000, 5, 248 CrossRef CAS.
- M. Kawai, P. Cras, P. Richey, M. Tabaton, D. E. Lowery and P. A Gonzalez-DeWhitt, et al. , Am. J. Pathol., 1992, 140, 947 Search PubMed.
- C. Tretiakoff, Contribution à l'étude de l'anatomie pathologique du locus niger de Sömmering avec quelques déductions relatives à la pathogénie des troubles du tonus musculaire et de la maladie de Parkinson. Medical Thesis, Paris: Jouve et Cie, 1919 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S6. See DOI: 10.1039/c0nr00345j |
| ‡ Present address: European Center for Nanomedicine, Neurological Institute “Carlo Besta”, IFOM-IEO-campus, Milan, Italy. |
| § Present address: NIHR Pancreatic Biomedical Unit at the Royal Liverpool University Hospital, Liverpool, UK. |
| This journal is © The Royal Society of Chemistry 2010 |