New hyperbranched polyaryleneethynylene containing azobenzenechromophore moieties in the main chain: facile synthesis, large optical nonlinearity and high thermal stability†
Zhong'an
Li
a,
Wenbo
Wu
a,
Cheng
Ye
b,
Jingui
Qin
a and
Zhen
Li
*a
aDepartment of Chemistry, Hubei Key Lab on Organic and Polymeric Opto-Electronic Materials, Wuhan University, Wuhan 430072, China. E-mail: lizhen@whu.edu.cn; Fax: +86-27-68756757; Tel: +86-27-62254108
bOrganic Solids Laboratories, Institute of Chemistry, The Chinese Academy of Sciences, Beijing 100080, China
First published on 16th December 2009
Abstract
A new main-chain NLO hyperbranched polymer (P1) was first prepared through a simple synthetic route, which demonstrated much better NLO properties (d33 value up to 143.8 pm/V) than its corresponding linear analogue (P2), since the three-dimensional spatial isolation from the highly branched structure could greatly enhance the poling efficiency.
Organic/polymeric nonlinear optical (NLO) materials have been put forth as promising candidates for future high technology applications such as frequency doublers, optical storage devices, and electro-optic (EO) switches and modulators.1–3 In the past decades, a major effort devoted in this area has been made to tackle the aggregation-quenching problem, caused by the strong intermolecular electrostatic interactions among high dipole moment chromophores, which directly made the required ordered noncentrosymmetric alignment of the chromophore moieties during the poling process under an electric field a daunting task.4,5 Fortunately, an elaborate molecular structure—three-dimensional (3D) was reported to minimize the dipole–dipole interactions by locking a chromophore inside the dendrimer shell, and thus enhance the macroscopic NLO activities by applying the site-isolation principle.4,6,7 As typical examples, many novel series of EO dendrimers were prepared.4,8,9
However, dendrimers usually require highly complicated and repetitive synthetic routes, resulting in the production of dendrimers being relatively costly. In sharp contrast, hyperbranched polymers could be typically obtained in a one-pot reaction, even in larger quantities.10 Similar to dendrimers, hyperbranched polymers are also spherical in their structure, and the three-dimensional spatial separation of the chromophore moieties endows them with favorable site isolation effects,11 while their void-rich topological structure could minimize optical loss in the NLO process.4b Therefore, the globular macromolecules are thus alternative promising candidates for NLO materials with large bulk EO activities. To date, the literatures concerned on hyperbranched NLO polymers were still scarce,12 and in our previous work, we have successfully prepared some hyperbranched NLO polymers with good comprehensive properties.13 But so far, the reported hyperbranched NLO polymers are side-chain ones, considering that the main-chain linear NLO polymer generally demonstrated stabilized oriented dipoles at elevated temperatures, and to further develop more hyperbranched NLO polymers with exciting properties, in this paper, we reported the synthesis and characterization of a new main-chain hb-PAE (P1) through a “A3 + B2” approach via a Sonogashira coupling reaction. For comparison, its corresponding linear analogue (P2) was also prepared. Thus, this mid-type NLO polymer was expected to possess both the merits of hyperbranched and main-chain NLO polymers: large optical nonlinearity and high stabilization of dipole moments.
As shown in Scheme 1, the synthetic route was very facile, and the experimental details were described in the ESI† . Chromophore1 underwent a typical “A3 + B2”-type of hyperbranched polymerization with aryl triiodide (2) to afford a hyperbranched poly(aryleneethynylene) (P1). The reaction was carefully carried out, to avoid it becoming out of control and yielding an insoluble network. Actually, during the reaction process, there were some red insoluble solid produced. However, after being purified by several precipitations of the THF solution into acetone, the yield of P1 was still satisfactory (61.5%). For comparison, the analogous linear polymer (P2) containing the same active chromophore moieties, was also prepared under the similar conditions (Scheme 2). It was easily seen that P1 and P2 were main-chain polymers, as most parts of the chromophore moieties were bonded into the polymeric backbone. The structure of P1 was different from those of the reported NLO hyperbranched polymers in our previous work or the literature, all of which were side-chain polymers.11,12 Thus, we expected that P1 with a new structure could merge the concepts of main-chain and hyperbranched polymers, and exhibit large NLO effects and high temporal stabilities.
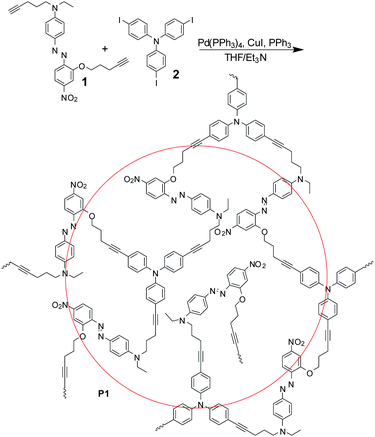 | ||
| Scheme 1 The synthetic route of hyperbranched polymerP1. | ||
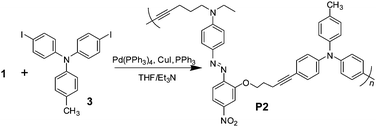 | ||
| Scheme 2 The synthetic route of linear polymer P2. | ||
Chromophore 1 and polymersP1 and P2 were characterized by spectroscopic methods, which gave satisfactory spectral data (see Experimental Section in ESI† for detailed analysis data). Fig. S1† shows the IR spectra of chromophore1 and polymersP1 and P2. After polymerization, the C≡CH stretching vibration (3290 cm−1) of chromophore3 disappeared, while those of the nitro groups at 1515 and 1337 cm−1 remained. No unexpected resonance peaks were observed in the 1H NMR spectra of the monomer and the polymers: all the peaks could be readily assigned to the resonances of appropriate protons as demonstrated in Scheme 1 and 2. It was easily seen that the resonance peak of the acetyleneprotons in chromophore1 at δ 2.00 ppm disappeared in the spectrum of P1, confirming the successful polymerization (Fig. S2 and 3, ESI† ). The two polymers were soluble in common organic solvents, the UV–vis absorption spectra of the chromophore and polymers were shown in Fig. S4 (ESI† ), with the maximum absorption wavelengths (λmax) for the π–π* transition of the azo moieties listed in the Experimental section and Table 1. The λmax wavelengths of polymers were nearly the same as those of chromophore1, indicating the surrounding environment of chromophore moieties did not change during the polymerization process. The molecular weights of polymers were determined by gel permeation chromatography (GPC) with THF as an eluent and polystyrene standards as calibration standards, and the results were summarized in Table 1. Their Mw values were very good, higher than those reported previously.13a
| No. | Yield (%) | M w a | M w/Mna | λ max b /nm | Tec /°C | l s d /μm | d 33 e (pm/V) | d 33 (∞) f (pm/V) | N g | Φ h |
|---|---|---|---|---|---|---|---|---|---|---|
| a Determined by GPC in THF on the basis of a polystyrene calibration. b The maximum absorption wavelength of polymer solutions in THF, while the maximum absorption wavelength of the corresponding small chromophore molecules in diluted THF solutions are given in the parentheses. c The best poling temperature. d Film thickness. e Second harmonic generation (SHG) coefficient. f The nonresonant d33 values calculated by using the approximate two-level model. g The effective chromophore loading density. h Order parameter Φ = 1−A1/A0, A1 and A0 are the absorbance of the polymer film after and before corona poling, respectively. | ||||||||||
| P1 | 61.5 | 11100 | 1.73 | 488 (489) | 180 | 0.21 | 143.8 | 15.1 | 0.51 | 0.21 |
| P2 | 66.7 | 11550 | 2.54 | 491 (489) | 145 | 0.30 | 52.9 | 5.6 | 0.54 | 0.10 |
P1 and P2 exhibited good film-forming ability, and their poled thin films were easily prepared to evaluate the NLO activity. The test method and conditions were shown in the ESI† . From the experimental data, the d33 values of P1 and P2 were calculated to be 143.8 and 52.9 pm/V, respectively, at the 1064 nm fundamental wavelength (Table 1). The obtained results were really very encouraging, and the d33 value of the hyperbranched polymerP1 was more than 2.7 times that of the linear polymer P2. Repeating the measurement gave similar results within experimental errors, verifying the data reproducibility. Moreover, in comparison with the d33 values of other linear NLO side-chain polymers bearing similar azobenzenechromophore moieties (including those with suitable isolation groups introduced),14 the d33 value of P1 (143.8 pm/V) was also outstandingly high. It was well-known that in theory,15 under identical experimental conditions, the d33 value was proportional to the density of the chromophore moieties (eqn S1, ESI† ). Thus, considering their same active D-π-A structure and similar effective chromophore density (0.51 for P1 and 0.54 for P2), the enhanced macroscopic NLO activities should be ascribed to the 3D architectural structure of the hyperbranched polymers, which could greatly improve the poling efficiency. Generally, due to their rigid structure, the main-chain NLO polymers demonstrated low poling efficiencies, and the effective oriented alignment of the dipole moments was very difficult to be realized.16 Thus, the improved efficiency of P1 would give a clue to address the above issues. Since there might be some resonant enhancement due to the absorption of the chromophore moieties at 532 nm, the d33(∞) values of polymers were calculated by using the approximate two-level model (Table 1). The d33(∞) value of P1 was still very high (15.1 pm/V), much larger than those of the reported linear NLO polymers, especially main-chain polymers, with nitro as the acceptor.13 To further study the alignment behavior of the chromophore moieties in the polymers, the order parameter (Φ) of the polymers was also measured and calculated from the change of the UV–vis spectra of their films before and after corona poling under an electric field (Fig. 1), according to the equation described in Table 1 (footnote h). The tested Φ values were in accordance with their d33 values: P1 exhibited a higher Φ value (0.21) than that of P2 (0.10), indicating much effective oriented alignment of the dipole moments alignment achieved in P1, further confirming the above conclusion.
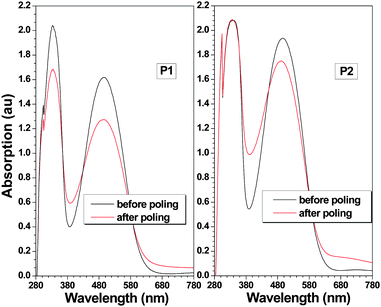 | ||
| Fig. 1 Absorption spectra of the film of P1 and P2 before and after poling. | ||
The dynamic thermal stabilities of the NLO activities of the polymers were investigated by depoling experiments, in which the real time decays of their SHG signals were monitored as the poled films were heated from 40 to 180 °C in air at a rate of 4 °C min−1. As shown in Fig. 2, the long-term temporal stability of the polymers was relatively good, the onset temperature for decays in P1 and P2 was found be as high as 153 and 119 °C, respectively, due to the rigid-rod structure of the main-chain. Thus, coupled with its much higher d33 value, P1 demonstrated both of the merits of hyperbranched and main-chain NLO polymers: large optical nonlinearity and high stabilization of dipole moments, as expected. In addition, the thermal curing of the polymer might also play an important role in boosting their resistance against the thermal depoling, since the acetylenic triple bonds could readily undergo thermal polymerization by moderate heating.17
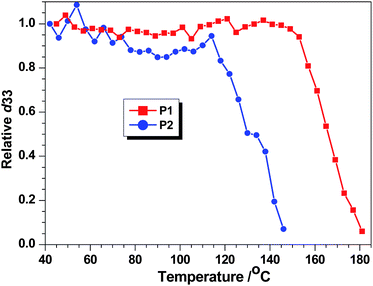 | ||
| Fig. 2 Decay curves of SHG coefficient of P1and P2 as a function of temperature. | ||
In this work, a new hyperbranched poly(aryleneethynylene)P1 was successfully prepared through an “A3 + B2” approach via a simple Sonogashira coupling reaction, in which the chromophore moieties were part of the polymer backbone, just like the linear main-chain polymers. P1 exhibited much better NLO properties (including a much larger d33 value, improved poling efficiency and enhanced temporal stability) than its corresponding linear analogue (P2), due to the three-dimensional spatial isolation from its highly branched structure, making it a promising candidate for optoelectronic applications. In addition, our results might provide a new solution to solve the “nonlinearity–stability trade off” existing in main-chain polymers to some degree.
We are grateful to the National Science Foundation of China (no. 20674059) and the program of NCET for financial support.
References
- (a) J. Zyss, Molecular Nonlinear Optics: Materials, Physics and Devices, Academic Press, Boston, 1994 Search PubMed; (b) L. R. Dalton, Chem. Ind., 1997, 7, 510; (c) T. J. Marks and M. A. Ratner, Angew. Chem., Int. Ed. Engl., 1995, 34, 155 CrossRef CAS; (d) K. A. Firestone, P. Reid, R. Lawson, S.-H. Jang and L. R. Dalton, Inorg. Chim. Acta, 2004, 357, 3957 CrossRef CAS.
- (a) S. R. Marder, B. Kippelen, A. K. Y. Jen and N. Peyghambarian, Nature, 1997, 388, 845 CrossRef CAS; (b) M. Lee, H. E. Katz, C. Erben, D. M. Gill, P. Gopalan, J. D. Heber and D. J. McGee, Science, 2002, 298, 1401 CrossRef CAS; (c) Y. Shi, C. Zhang, H. Zhang, J. H. Bechtel, L. R. Dalton, B. H. Robinson, W. H. Steier and L. R. Dalton, Science, 2000, 288, 119 CrossRef CAS; (d) H. Chen, B. Chen, D. Huang, D. Jin, J. Luo, A. K.-Y. Jen and R. Dinu, Appl. Phys. Lett., 2008, 93, 043507 CrossRef.
- (a) D. M. Burland, R. D. Miller and C. A. Walsh, Chem. Rev., 1994, 94, 31 CrossRef CAS; (b) Q. Wang, L. M. Wang and L. P. Yu, Macromol. Rapid Commun., 2000, 21, 723 CrossRef CAS; (c) F. Kajzar, K.-S. Lee and A. K.-Y. Jen, Adv. Polym. Sci., 2003, 161, 1 CAS.
- (a) M. J. Cho, D. H. Choia, P. A. Sullivan, A. J.-P. Akelaitis and L. R. Dalton, Prog. Polym. Sci., 2008, 33, 1013 CrossRef CAS; (b) H. Ma, S. Liu, J. Luo, S. Suresh, L. Liu, S. H. Kang, M. Haller, T. Sassa, L. R. Dalton and A. K.-Y. Jen, Adv. Funct. Mater., 2002, 12, 565 CrossRef CAS.
- (a) A. W. Harper, S. Sun, L. R. Dalton, S. M. Garner, A. Chen, S. Kulluri, W. H. Steier and B. H. Robinson, J. Opt. Soc. Am. B, 1998, 15, 329 Search PubMed; (b) B. H. Robinson and L. R. Dalton, J. Phys. Chem. A, 2000, 104, 4785 CrossRef CAS; (c) B. H. Robinson, L. R. Dalton, H. W. Harper, A. Ren, F. Wang, C. Zhang, G. Todorova, M. Lee, R. Aniszfeld, S. Garner, A. Chen, W. H. Steier, S. Houbrecht, A. Persoons, I. Ledoux, J. Zyss and A. K.-Y. Jen, Chem. Phys., 1999, 245, 35 CrossRef CAS.
- (a) P. A. Sullivan, A. J.-P. Akelaitis, S. K. Lee, G. McGrew, S. K. Lee, D. H. Choi and L. R. Dalton, Chem. Mater., 2006, 18, 344 CrossRef CAS; (b) S. Yokoyama, T. Nakahama, A. Otomo and S. Mashiko, J. Am. Chem. Soc., 2000, 122, 3174 CrossRef CAS; (c) P. Gopalan, H. E. Katz, D. J. McGee, C. Erben, T. Zielinski, D. Bousquet, D. Muller, J. Grazul and Y. Olsson, J. Am. Chem. Soc., 2004, 126, 1741 CAS; (d) J. Y. Do, S. K. P. ark, J. J. Ju, S. Park and M. Lee, Macromol. Chem. Phys., 2003, 204, 410 CrossRef CAS; (e) J. Y. Do and J. J. Ju, Macromol. Chem. Phys., 2005, 206, 1326 CrossRef CAS.
- (a) P. Busson, J. Örtegren, H. Ihre, U. W. Gedde, A. Hult, G. Andersson, A. Eriksson and M. Lindgren, Macromolecules, 2002, 35, 1663 CrossRef CAS; (b) O. Varnavski, A. Leanov, L. Liu, J. Takacs and T. Goodson, III, J. Phys. Chem. B, 2000, 104, 179 CrossRef CAS; (c) J. Gao, Y. Cui, J. Yu, Z. Wang, M. Wang, J. Qiu and G. Qian, Macromolecules, 2009, 42, 2198 CrossRef CAS; (d) Z. Li, G. Yu, W. Wu, Y. Liu, C. Ye, J. Qin and Z. Li, Macromolecules, 2009, 42, 3864 CrossRef CAS.
- (a) M. Fischer and F. Vögtle, Angew. Chem., Int. Ed., 1999, 38, 884 CrossRef; (b) A.W. Bosman, H.M. Janssen and E.W. Meijer, Chem. Rev., 1999, 99, 1665 CrossRef CAS; (c) S.-E. Stiriba, H. Frey and R. Haag, Angew. Chem., Int. Ed., 2002, 41, 1329 CrossRef CAS; (d) J. M. J. Fréchet, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3713 CrossRef CAS; (e) F. Zeng and S. C. Zimmerman, Chem. Rev., 1997, 97, 1681 CrossRef CAS.
- (a) W. Shi, J. Luo, S. Huang, X.-H. Zhou, T.-D. Kim, Y.-J. Cheng, B. M. Polishak, T. R. Younkin, B. A. Block and A. K.-Y. Jen, Chem. Mater., 2008, 20, 6372 CrossRef CAS; (b) H. Ma and A. K. Y. Jen, Adv. Mater., 2001, 13, 1201 CrossRef CAS; (c) T.-D. Kim, J.-W. Kang, J. Luo, S.-H. Jang, J.-W. Ka, N. Tucker, J. B. Benedict, L. R. Dalton, T. Gray, R. M. Overney, D. H. Park, W. N. Herman and A. K.-Y. Jen, J. Am. Chem. Soc., 2007, 129, 488 CrossRef CAS.
- (a) Y. H. Kim, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 1685 CrossRef CAS; (b) A. Sunder, J. Heinemann and H. Frey, Chem.–Eur. J., 2000, 6, 2499 CrossRef CAS; (c) M. Häußer and B. Z. Tang, Adv. Polym. Sci., 2007, 209, 1; (d) B. Voit, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2505 CrossRef CAS; (e) M. Scholl, Z. Kadlecova and H.-A. Klok, Prog. Polym. Sci., 2009, 34, 24 CrossRef CAS.
- (a) J. M. J. Fréchet, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4782 CrossRef CAS; (b) S. Hecht and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2001, 40, 74 CrossRef CAS; (c) J. M. J. Fréchet, M. Henmi, I. Gitsov, S. Aoshima, M. R. Leduc and R. B. Grubbs, Science, 1995, 269, 1080 CrossRef CAS.
- (a) Y. Bai, N. Song, J. P. Gao, X. Sun, X. Wang, G. Yu and Z. Y. Wang, J. Am. Chem. Soc., 2005, 127, 2060 CrossRef CAS; (b) J. Xie, X. Deng, Z. Cao, S. Q. hen, W. Zhang and W. Shi, Polymer, 2007, 48, 5988 CrossRef CAS; (c) H.-L. Chang, T.-Y. Chao, C.-C. Yang, S. A. Dai and R.-J. Jeng, Eur. Polym. J., 2007, 43, 3988 CrossRef CAS; (d) J. Xie, L. Hu, W. Shi, X. Deng, Z. Cao and Q. Shen, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 1140 CrossRef CAS.
- (a) Z. Li, A. Qin, J. W. Y. Lam, Y. Dong, Y. Dong, C. Ye, I. D. Williams and B. Z. Tang, Macromolecules, 2006, 39, 1436 CrossRef CAS; (b) Z. Zhu, Z. Li, Y. Tan, Z. Li, Q. Li, Q. Zeng, C. Ye and J. Qin, Polymer, 2006, 47, 7881 CrossRef CAS; (c) Z. Li, G. Yu, P. Hu, C. Ye, Y. Liu, J. Qin and Z. Li, Macromolecules, 2009, 42, 1589 CrossRef CAS.
- (a) Z. Li, Z. Li, C. Di, Z. Zhu, Q. Li, Q. Zeng, K. Zhang, Y. Liu, C. Ye and J. Qin, Macromolecules, 2006, 39, 6951 CrossRef CAS; (b) Z. Li, Q. Zeng, Z. Li, S. Dong, Z. Zhu, Q. Li, C. Ye, C. Di, Y. Liu and J. Qin, Macromolecules, 2006, 39, 8544 CrossRef CAS; (c) Q. Zeng, Z. Li, Z. Li, C. Ye, J. Qin and B. Z. Tang, Macromolecules, 2007, 40, 5634 CrossRef CAS; (d) Z. Li, S. Dong, G. Yu, Z. Li, Y. Liu, C. Ye and J. Qin, Polymer, 2007, 48, 5520 CrossRef CAS; (e) Z. Li, G. Yu, S. Dong, W. Wu, Y. Liu, C. Ye, J. Qin and Z. Li, Polymer, 2009, 50, 2806 CrossRef CAS.
- C. R. Moylan, R. D. Miller, R. J. Twieg, V. Y. Lee, I. H. McComb, S. Ermer, S. M. Lovejoy and D. S. Leung, Proc. SPIE–Int. Soc. Opt. Eng., 1995, 2527, 150 CAS.
- (a) W. Kohler, D. R. Robello, C. S. Willand and D. J. Willams, Macromolecules, 1991, 24, 4589 CrossRef; (b) Y. Zhang, L. Wang, T. Wada and H. Sasabe, Macromolecules, 1996, 29, 1569 CrossRef CAS; (c) W. J. Kuo, G. H. Hsiue and R. J. Jeng, Macromol. Rapid Commun., 2001, 22, 601 CrossRef CAS.
- (a) H. C. Dong, R. H. Zheng, J. W. Y. Lam, M. Häussler and B. Z. Tang, Macromolecules, 2005, 38, 6382 CrossRef CAS; (b) Z. Li, Y. Q. Dong, A. Qin, J. W. Y. Lam, Y. P. Dong, W. Yuan, J. Sun, J. Hua, K. S. Wong and B. Z. Tang, Macromolecules, 2006, 39, 467 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: Detailed synthetic procedures and characterization data for the monomers and polymers; figures of FT-IR spectra, 1H NMR spectra, UV–vis spectra. See DOI: 10.1039/b9py00225a/. |
| This journal is © The Royal Society of Chemistry 2010 |