Sequential double click reactions: a highly efficient post-functionalization method for optoelectronic polymers†‡
Yongrong
Li
a and
Tsuyoshi
Michinobu
*bc
aDepartment of Organic and Polymeric Materials, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8552, Japan
bGlobal Edge Institute, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8550, Japan. E-mail: michinobu.t.aa@m.titech.ac.jp; Fax: +81-3-5734-3774; Tel: +81-3-5734-3774
cPRESTO, Japan Science and Technology Agency (JST), Kawaguchi, Saitama, Japan
First published on 11th December 2009
Abstract
A new post-functionalization method based on the Huisgen's 1,3-dipolar cycloaddition between azides and alkynes, followed by the atom-economic addition reaction between electron-rich alkynes and tetracyanoethylene (TCNE) was developed to introduce the donor–acceptor chromophores into polystyrene derivatives in remarkably high yields.
The new concept of click chemistry has attracted widespread attention in polymer science.1 The Cu(I)-catalyzed azide-alkyne cycloaddition (CuAAC) is one of the most frequently employed reactions, which is capable of selectively combining two components in terms of covalent bonds in excellent yields.2 In order to expand the applicability of this concept, other click chemistry-type reactions, such as the Diels–Alder reaction3 and thiol-ene reaction,4 have also been developed and their utility in polymer chemistry was demonstrated by application to post-functionalizations. However, these click reactions do not have specific advantages regarding applications to conjugated systems due to the disruption of effective conjugation length and the resulting poor optoelectronic properties of the products.5
Intramolecular donor–acceptor interactions often produce optoelectronically useful chromophores. Recently, we reported the efficient introduction of nonplanar donor–acceptor chromophores, donor-substituted 1,1,4,4-tetracyanobuta-1,3-dienes (TCBDs), in the main chains6 and side chains7 of aromaticpolymers by high-yielding addition reactions between electron-rich alkynes and a strong acceptor, tetracyanoethylene (TCNE), under mild conditions. In particular, the TCNE addition to the polymer side chains was quantitative at room temperature, and accordingly, this reaction8 satisfies most of the requirements for click chemistry, as suggested in the original review.2 Additional advantages of this reaction over the conventional click reactions are that no metal catalysts are necessary and the products feature strong charge-transfer (CT) bands in the visible (and near-IR) region, potent redox activities, and related optoelectronic properties. For example, the excellent second-order9 and third-order10 nonlinear optical effects have already been reported for the small and dendritic molecular architectures.11
In this Communication, we report the first successful introduction of this donor–acceptor chromophore into polystyrene side chains using the double click reactions based on the CuAAC and TCNE addition. The double click reactions of aliphatic polymers have in the past been investigated,12 but it is very rare to realize optoelectronic polymers.
Poly(4-azidomethylstyrene)
1
, prepared from poly(4-chloromethlystyrene) and sodium azide, was employed as the precursor polymer.13 The molecular weight (Mn) and the polydispersity (Mw/Mn) were 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 and 1.75, respectively. The azide groups were reacted with the terminal alkynes of 2p or 2m in the presence of Cu(I) catalysts, yielding the tolane-appended polymers3p and 3m (Scheme 1). Note that 2p and 2m possess both terminal and internal alkynes, and the terminal alkynes selectively react under the CuAAC conditions. Gratifyingly, we found that when the reaction is performed in DMF, the resulting high molecular weight polymers are deposited from the solutions. Thus, a simple filtration of the deposited precipitates after the reaction for 24 h provided the desired polymers3p and 3m in the fairly good yield of 84%. Evaporation of the filtrate and the subsequent precipitation into a water–methanol (4 : 1) mixture afforded the residual desired polymers. The combined yields amounted to 98%. The GPC measurements revealed a reasonable molecular weight increase (Fig. 1). The Mn and Mw/Mn values are 52300 and 1.55 for 3p and 49000 and 1.85 for 3m, respectively. The slightly lower molecular weight and higher polydispersity of the m-phenylene-linked 3m as compared to the p-phenylene-linked 3p are supposed to reflect the different hydrodynamic volumes in the GPC measurements. The 1H NMR spectra confirmed the chemical structures of both polymers (Fig. 2). The benzyl proton peak of 1 at 4.60 ppm completely shifted to 5.47 ppm for 3p and 5.30 ppm for 3m. Moreover, the dialkylaniline peaks appeared after the click modification. In the IR spectra of 3p and 3m, the vibrational peak at 2095 cm−1 ascribed to the azide groups of 1 disappeared, whereas the alkyne vibrational peak at 2207 cm−1 remained. These results are consistent with the quantitative addition, ensuring the use of CuAAC in click chemistry reactions.
500 and 1.75, respectively. The azide groups were reacted with the terminal alkynes of 2p or 2m in the presence of Cu(I) catalysts, yielding the tolane-appended polymers3p and 3m (Scheme 1). Note that 2p and 2m possess both terminal and internal alkynes, and the terminal alkynes selectively react under the CuAAC conditions. Gratifyingly, we found that when the reaction is performed in DMF, the resulting high molecular weight polymers are deposited from the solutions. Thus, a simple filtration of the deposited precipitates after the reaction for 24 h provided the desired polymers3p and 3m in the fairly good yield of 84%. Evaporation of the filtrate and the subsequent precipitation into a water–methanol (4 : 1) mixture afforded the residual desired polymers. The combined yields amounted to 98%. The GPC measurements revealed a reasonable molecular weight increase (Fig. 1). The Mn and Mw/Mn values are 52300 and 1.55 for 3p and 49000 and 1.85 for 3m, respectively. The slightly lower molecular weight and higher polydispersity of the m-phenylene-linked 3m as compared to the p-phenylene-linked 3p are supposed to reflect the different hydrodynamic volumes in the GPC measurements. The 1H NMR spectra confirmed the chemical structures of both polymers (Fig. 2). The benzyl proton peak of 1 at 4.60 ppm completely shifted to 5.47 ppm for 3p and 5.30 ppm for 3m. Moreover, the dialkylaniline peaks appeared after the click modification. In the IR spectra of 3p and 3m, the vibrational peak at 2095 cm−1 ascribed to the azide groups of 1 disappeared, whereas the alkyne vibrational peak at 2207 cm−1 remained. These results are consistent with the quantitative addition, ensuring the use of CuAAC in click chemistry reactions.
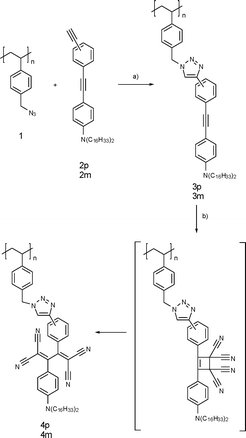 | ||
| Scheme 1 Sequential click post-functionalization of polystyrene derivative 1. (a) CuSO4·5H2O, sodium ascorbate, DMF; (b) TCNE, 1,2-dichloroethane. | ||
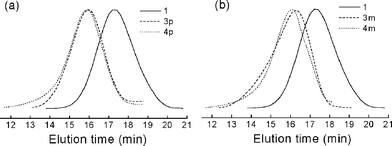 | ||
| Fig. 1 GPC profiles of (a) 1, 3p, and 4p and (b) 1, 3m, and 4m. | ||
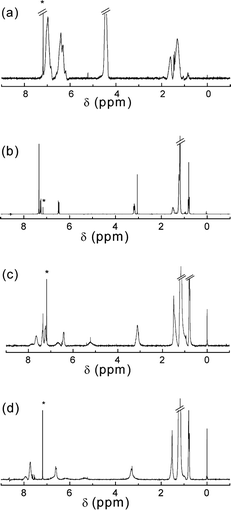 | ||
| Fig. 2 1H NMR spectra of (a) 1, (b) 2p, (c) 3p and (d) 4p in CDCl3 at 20 °C. | ||
Subsequently, the TCNE addition to the polymers3p and 3m were examined. The titration experiments of 3p and 3m with TCNE were monitored by UV/Vis spectroscopy. New CT bands appeared, and the intensities increased with the increasing amount of the added TCNE, finally leading to completely red solutions (Fig. 3). The presence of the isosbestic points at 308 and 382 nm for 3p and at 306 and 377 nm for 3m indicates no side reactions during the TCNEclick reactions. The positions of the most intense CT bands (480.5 nm for 3p and 483 nm for 3m) were consistent with the monomeric TCBD structures8 and did not change during the experiments, suggesting negligible interactions between the chromophores.
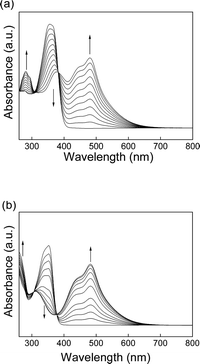 | ||
| Fig. 3 UV/Vis spectral changes of (a) 3p and (b) 3m upon titration with TCNE (0–1.0 equiv.) in chloroform at 20 °C. | ||
The UV/Vis spectral results enabled us to prepare the full TCNEadduct polymers. The addition of the stoichiometric amount of TCNE, followed by evaporation of the solvents, quantitatively furnished the desired TCBD polymers, 4p and 4m, without any purification processes (Scheme 1). The molecular weights of the TCBD polymers were slightly increased from the precursor polymers, as shown in the GPC profiles (Fig. 1). Thus, the Mn values of 4p and 4m were 58500 and 51000, respectively. The polydispersities were almost similar to those of the precursors. In the 1H NMR spectra, some peaks, such as the benzyl protons, became broader as compared to the precursors, implying the increased rotational barriers of part of the single bonds due to the bulky and twisted TCBD moieties (Fig. 2). The IR spectra also substantiated the chemical structures. A weak alkyne vibrational peak of the precursor tolane polymers was replaced by a strong cyano one at 2214 cm−1. Moreover, the elemental analysis results of 4p and 4m showed a good agreement with the calculated values. All these results support the perfect post-functionalization by the alkyne-TCNEclick reactions.
In order to investigate the importance of the reaction order, we also tried the other double click reactions based on the first TCNE addition to 2p and 2m, followed by the CuAAC addition to 1 (Scheme 2). The TCNE addition to 2p and 2m quantitatively afforded the corresponding TCBD molecules, 5p and 5m, at room temperature. The terminal alkynes of 5p and 5m were chemically stable and therefore subjected to the reaction with 1 in the presence of Cu(I) catalysts. The resulting polymers, 6p and 6m, showed absorption spectral shapes similar to 4p and 4m, respectively, but the peak instensities were apparently lower (Fig. S1‡ ). Furthermore, the Mn values of 6p (42800) and 6m (44800) were lower than those of 4p and 4m, and the polydispersities were much higher (Table S1‡ ). All these results indicate that it is difficult to densely attach the bulky TCBD chromophores as the polystyrene side chain. Accordingly, the original route based on the first incorporation of the linear tolane side chains by CuAAC, followed by the TCNE addition is a more efficient method to construct the high density donor–acceptor side chains. Based on the measured molecular weights, the reaction order is more significant for the p-linked derivative than the m-linked analogue.
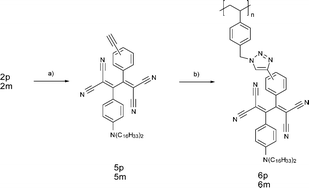 | ||
| Scheme 2 Post-functionalization by TCNEclick reactions followed by CuAAC. (a) TCNE, 1,2-dichloroethane; (b) 1, CuSO4·5H2O, sodium ascorbate, DMF. | ||
Another important feature of the donor–acceptor systems is the redox activities. The tolane polymers, 3p and 3m, displayed the only aniline-centered, quasi-reversible oxidation waves at 0.28 and 0.17 V (vs. Fc/Fc+), respectively, in the cyclic voltammograms (CVs) recorded in CH2Cl2 (+0.1 M (nC4H9)4NClO4) at 20 °C (Table S2 and Fig. S2‡ ). On the other hand, the TCBD polymers, 4p and 4m, showed anodically shifted oxdation potentials at 0.84 and 0.73 V, respectively, as well as the TCBD-centered reduction potentials at −0.91 and −0.98 V, respectively. In contrast to the small TCBD molecules showing two well-resolved reduction steps,8 the polymers displayed only the single reduction waves. The electrochemical band gaps of both 4p and 4m, calculated from the oxidation and reduction potentials, are ∼1.7 V. This value is comparable to the optical band gaps determined by the end absorptions (1.72 eV).
In conclusion, we have developed a highly efficient post-functionalization method using two different click reactions, which can be applicable to various polymers. The importance of the reaction order was rationally explained by the steric reasons. It was found that the alkyne-TCNEclick reactions proceeds even in sterically congested environments and provides the potent donor–acceptor chromophores in excellent yields. All the obtained polymers were thermally stable with decomposition temperatures exceeding 340 °C (Table S1‡ ). Combined with the high thermal stability, they are promising for a variety of optoelectronic device applications, which are our future projects.
This work was supported by a Grant-in-Aid for Scientific Research and the Special Coordination Funds for Promoting Science and Technology from MEXT, Japan.
Notes and references
- (a) D. Fournier, R. Hoogenboom and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1369 RSC; (b) M. Meldal and C. W. Tomøe, Chem. Rev., 2008, 108, 2952 CrossRef CAS; (c) B. Z. Tang, Macromol. Chem. Phys., 2008, 209, 1303 CrossRef CAS; (d) W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2008, 29, 952 CrossRef CAS; (e) P. Lundberg, C. J. Hawker, A. Hult and M. Malkoch, Macromol. Rapid Commun., 2008, 29, 998 CrossRef CAS; (f) J. A. Johnson, M. G. Finn, J. T. Koberstein and N. J. Turro, Macromol. Rapid Commun., 2008, 29, 1052 CrossRef CAS; (g) C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900 CrossRef CAS; (h) C. Barner-Kowollik and A. J. Inglis, Macromol. Chem. Phys., 2009, 210, 987 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- (a) B. Gacal, H. Durmaz, M. A. Tasdelen, G. Hizal, U. Tunca, Y. Yagci and A. L. Demirel, Macromolecules, 2006, 39, 5330 CrossRef CAS; (b) K. Ishida and N. Yoshie, Macromol. Biosci., 2008, 8, 916 CrossRef CAS.
- K. L. Killops, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 5062 CrossRef CAS.
- (a) D. J. V. C. van Steenis, O. R. P. David, G. P. F. van Strijdonck, J. H. van Maarseveen and J. N. H. Reek, Chem. Commun., 2005, 4333 RSC; (b) M. A. Karim, Y.-R. Cho, J. S. Park, S. C. Kim, H. J. Kim, J. W. Lee, Y.-S. Gal and S.-H. Jin, Chem. Commun., 2008, 1929 RSC.
- T. Michinobu, H. Kumazawa, K. Noguchi and K. Shigehara, Macromolecules, 2009, 42, 5903 CrossRef CAS.
- T. Michinobu, J. Am. Chem. Soc., 2008, 130, 14074 CrossRef CAS.
- (a) T. Michinobu, J. C. May, J. H. Lim, C. Boudon, J.-P. Gisselbrecht, P. Seiler, M. Gross, I. Biaggio and F. Diederich, Chem. Commun., 2005, 737 RSC; (b) T. Michinobu, C. Boudon, J.-P. Gisselbrecht, P. Seiler, B. Frank, N. N. P. Moonen, M. Gross and F. Diederich, Chem.–Eur. J., 2006, 12, 1889 CrossRef CAS; (c) M. Kivala and F. Diederich, Acc. Chem. Res., 2009, 42, 235 CrossRef CAS.
- (a) X. Wu, J. Wu, Y. Liu and A. K.-Y. Jen, J. Am. Chem. Soc., 1999, 121, 472 CrossRef CAS; (b) C. Cai, I. Liakatas, M.-S. Wong, M. Bösch, C. Bosshard, P. Günter, S. Concilio, N. Tirelli and U. W. Suter, Org. Lett., 1999, 1, 1847 CrossRef CAS; (c) H. Ma, B. Chen, T. Sassa, L. R. Dalton and A. K.-Y. Jen, J. Am. Chem. Soc., 2001, 123, 986 CrossRef CAS.
- (a) B. Esembeson, M. L. Scimeca, T. Michinobu, F. Diederich and I. Biaggio, Adv. Mater., 2008, 20, 4584 CrossRef CAS; (b) C. Koos, P. Vorreau, T. Vallaitis, P. Dumon, W. Bogaerts, R. Baets, B. Esembeson, I. Biaggio, T. Michinobu, F. Diederich, W. Freude and J. Leuthold, Nat. Photonics, 2009, 3, 216 Search PubMed.
- (a) M. Kivala, C. Boudon, J.-P. Gisselbrecht, P. Seiler, M. Gross and F. Diederich, Angew. Chem., Int. Ed., 2007, 46, 6357 CrossRef CAS; (b) M. Kivala, T. Stanoeva, T. Michinobu, B. Frank, G. Gescheidt and F. Diederich, Chem.–Eur. J., 2008, 14, 7638 CrossRef CAS.
- (a) B. Gacal, H. Akat, D. K. Balta, N. Arsu and Y. Yagci, Macromolecules, 2008, 41, 2401 CrossRef CAS; (b) A. Dag, H. Durmaz, E. Demir, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6969 CrossRef CAS; (c) H. Durmaz, A. Dag, A. Hizal, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7091 CrossRef CAS; (d) C. A. DeForest, B. D. Polizzotti and K. S. Anseth, Nat. Mater., 2009, 8, 659 CrossRef CAS; (e) L. Nurmi, J. Lindqvist, R. Randev, J. Syrett and D. M. Haddleton, Chem. Commun., 2009, 2727 RSC; (f) A. Dag, H. Durmaz, U. Tunca and G. Hizal, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 178 CrossRef CAS.
- The azide content in the polymer was determined to be 96% by the elemental analysis.
Footnotes |
| † All new molecules and polymers were fully characterized by IR, UV/Vis, 1H and 13C NMR, mass spectrometry or GPC and elemental analysis. |
| ‡ Electronic supplementary information (ESI) available: Synthetic procedures, polymer analyses, UV-Vis spectra, and electrochemistry results. See DOI: 10.1039/b9py00238c |
| This journal is © The Royal Society of Chemistry 2010 |