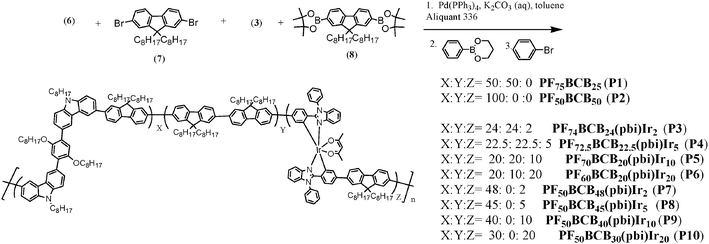
DOI:
10.1039/B9PY00276F
(Paper)
Polym. Chem., 2010,
1, 494-505
Electroluminescent main-chain copolymers containing phosphorescent benzimidazole-based iridium complexes as copolymerization backbone units or dopants
Received
3rd October 2009
, Accepted 5th November 2009
First published on 12th January 2010
Abstract
A series of novel electroluminescent main-chain copolymers containing fluorene-1,4-bis(9-octyl-9H-carbarzol-3yl)-2,5-dioctyloxy-benzene (BCB) segments and phosphorescent benzimidazole-based iridium (Ir) complexes in the backbones were synthesized by Suzuki coupling reaction. The relative intensity of phosphorescence and fluorescence were affected by the energy transfer and back transfer efficiencies between the polymer backbones and the iridium units as evidenced by solid state PL and EL spectra. PLED devices with a configuration of ITO/PEDOT: PSS (70 nm)/Ir-copolymers (P3–P10) or Ir-doped copolymers (P1–P2 doped with Ir-complex 4) (60–80 nm)/TPBI (40 nm)/LiF (1 nm)/Al (120 nm) were fabricated, where electroluminescence (EL) efficiencies depended on the chemical constituents and the triplet energies of the copolymers. PLED devices based on Ir-containing copolymerP8 or copolymerP2 doped with 5 mol% Ir-complex 4 exhibited white-light emissions with EL properties of ηext,max = 0.93%, ηc,max = 1.88 cd A−1, and Lmax = 1960 cd m−2 from the former (P8), and ηext,max = 4.09%, ηc,max = 10.94 cd A−1, and Lmax = 4870 cd m−2 from the latter (P2 doped with Ir-complex 4), respectively. The CIE coordinates, color-rendering index (CRI) and correlated color temperature (CCT) of the two PLED devices were (0.33, 0.30) at 13 V, 74 and 5966 K for the former, and (0.35, 0.32) at 15 V, 82 and 6147 K for the latter, respectively.
Introduction
Tremendous progress has been made on organic light-emitting diodes (OLEDs) after Tang and Van Slyke's1 and Burroughes et al.'s2 reports on LEDs consisting of small organic molecules and polymers, respectively. The development of phosphorescent transition metal complexes brought the external quantum efficiency (EQE) of OLEDs to a new milestone, because both singlet and triplet excitons can be harvested and thus an overall 100% internal quantum efficiency is possible.3 Many metal-based phosphorescent emitters have been developed in the past decade. Among these phosphorescent emitters, iridium,4platinum,5osmium,6 and ruthenium7 complexes have received considerable attention. These complexes were successfully used in polymer light-emitting diodes (PLEDs) and OLEDs which were usually prepared by solution process8 and vacuum deposition,9 respectively.
Compared with small molecules, polymers are more suitable for spin-coating or ink-jet techniques which renders flexible substrates and large area displays in conjunction with PLEDs.10 Nevertheless, the efficiencies of PLEDs fabricated viaspin-coating are generally lower than those of small molecule-based multi-layer devices fabricated viavacuum-deposition. To compensate for the lower efficiencies of PLEDs, iridium complexes were frequently doped into a polymer matrix11 or directly tethered to the polymer main chains,12 side chains,13 or terminals.14 For example, Gong et al. reported a highly efficient yellow-green electrophosphorescent OLED fabricated by doping tris(9,9-dihexyl-2-(pyridinyl-2′)fluorene) iridium(III) (Ir(DPF)3) into poly(N-vinylcarbazole) (PVK) blended with the electron-transporting 2-(4-biphenylyl)-5-(4-tert-butylphenyl)-1,3,4-oxadiazole (PBD), where an EQE of 10% and luminous efficiency (ηc) of 36 cd A−1 were achieved.11a Similarly, PVK doped with blue-emitting FIrpic, green-emitting Ir(ppy)3, and red-emitting (btp)2Ir(acac) complexes exhibited maximum EQE values at 1.3%, 5.1%, and 2.0%, respectively.11b
Alternatively, iridium complexes can be tethered to the backbones or side chains of polymers. Among PLEDs, fluorene units and their derivatives have been popularly used because of several advantages: high photoluminescence quantum yield (PLQY); excellent chemical, thermal, and photo stabilities; good solubilities and film-forming properties. Moreover, high-yielding synthetic routes for well-defined and high-molecular-weight polymers are readily available.15 Chen et al. grafted both carbazole and iridium complex moieties to polyfluorenes (PFs) via a saturated hydrocarbon spacer tethered at the carbon-9 position of the fluorene unit.13a The PLED device containing 1.3 mol% (btp)2Ir(acac) was reported to have an EQE of 1.59% and a power efficiency (ηp) of 2.8 cd A−1 at 7 V with a luminance of 65 cd m−2. A red-emitting 2-(1-naphthalene)pyridine-bicycloiridium/fluorenecopolymer developed by Zhen et al. was reported to have an EQE of 6.5% at a current density of 38 mA cm−2, and a luminance of 926 cd m−2 with an emission peak at 630 nm.12aPolymers synthesized by Jiang et al. had a backbone of fluorene-alt-carbazole with iridium complexes as side groups.13b The PLED devices fabricated from the previous polymers had the highest EQE of 4.9%, the luminous efficiency of 4.0 cd A−1 with 240 cd m−2 at 7.7 V, and the peak emission at 610 nm. Fluorene-alt-carbazolecopolymers P(F-alt-Cz)s developed by Zhang et al. had cyclometalated iridium fragments ligated to the β-diketone units of the polymer backbones. All fabricated PLED devices were almost free of efficiency decay even at high current density.12b Cao and co-workers,16 Shu and co-workers,17 and Lee et al.18 demonstrated that white light-emitting OLEDs (WOLEDs) were also possible by incorporation of a blue-emitting fluorene-based copolymer with green and red fluorescent (or phosphorescent) emitters in the backbones and side chains, respectively, where high current efficiencies of 6.1 cd A−1 and 4.8 cd A−1 were demonstrated correspondingly. It was interesting to note that most of the iridium complexes tethered to the conjugated copolymers were red-emitters.
A series of highly phosphorescent cyclometalated iridium (Ir) complexes containing benzimidazole-based ligands have previously been developed.19 High performance yellow- and green-emitting OLEDs have been achieved by using these complexes as guest emitters. Later, these Ir-complexes were successfully tethered with dendrons and efficient dendrimer-type LEDs (DLEDs) were fabricated.20 In view of the aforementioned easy solution-processing of polymers, and handy co-polymerization approach, we decided to extend benzimidazole-based cyclometalated Ir complexes to the polymer systems for possible applications in PLEDs including WOLEDs. For a fair comparison, the iridium units were tethered to the polymers backbones (Ir-copolymers) or doped into the analogous metal-free polymers (Ir-doped copolymers). In this study, we prepared a series of novel PLEDcopolymers consisting of fluorescent 1,4-bis(9-octyl-9H-carbazol-3yl)-2,5-dioctyloxy-benzene (BCB), fluorene, and phosphorescent benzimidazole-based Ir-complex units. Analogous metal-free polymers were prepared similarly except that the benzimidazole-based Ir-complex units were omitted. The physical and electroluminescent properties of these PLEDcopolymers were also investigated.
Results and discussion
Synthesis and characterization
The synthetic routes and chemical structures of the copolymers are shown in Schemes 1–3. The Ir-complexes 3 and 4 were synthesized by following the procedures of their iridium congener, (pbi)2Ir(acac)19 (the structure of (pbi)2Ir(acac) is shown Fig. 2(a)). The carbazole-based building block of the polymers, 1,4-bis(6-bromo-9-octyl-9H-carbarzol-3yl)-2,5-dioctyloxy-benzene (BCB-2Br, 6), was prepared by two successive steps: (1) Suzuki coupling of 9-octyl-9H-carbazol-3-ylboronic acid with 1,4-dibromo-2,5-bis(octyloxy)benzene to provide BCB; (2) bromination of BCB by using N-bromosuccinimide. Two octyloxy substituents were incorporated in BCB, where the carbazole segments were introduced to enhance the triplet energy in the polymers. The 2,5-bis(octyloxy)benzene unit in BCB was to improve the solubility and film-forming property of the desired copolymers. Copolymerization of monomers dibromo-substituted Ir-complex 3, BCB-2Br (6), and fluorenes (compounds 7 and 8) was achieved via Suzuki coupling reaction. The feed ratio of the iridium units was controlled at a level of 2, 5, 10, or 20 mol%. All polymers were characterized by 1H NMR spectroscopy, gel permeation chromatography (GPC), and elemental analysis. Actual compositions of the copolymers (characterized by 1H NMR) and the feed ratios of the monomers are listed in Table 1. The number-average molecular weights (Mn) of these copolymers lay in the range of 5000 to 20500 g mol−1 with a polydispersity index (PDI) ranging from 1.53 to 2.08.
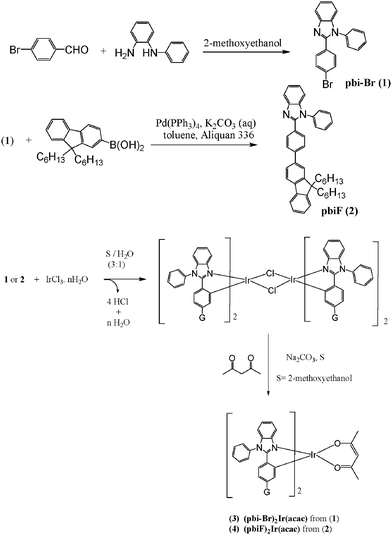 |
| | Scheme 1 Synthesis of bi ligands (1 and 2) and Ir-complexes 3 and 4. | |
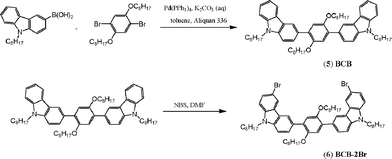 |
| | Scheme 2 Synthesis of monomer BCB-2Br (6) | |
|
Copolymer
|
Mn
|
Mw
|
PDI a |
Monomer feed ratio bF: BCB: Ir |
Monomer composition observed c |
Molecular weights were determined by GPC using polystyrene standards.
The feed ratio of F![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :BCB:iridium complex.
The iridium contents (F:BCB:iridium unit) in copolymers were estimated by 1H NMR. :BCB:iridium complex.
The iridium contents (F:BCB:iridium unit) in copolymers were estimated by 1H NMR.
|
|
P1
|
20500 |
33900 |
1.65 |
75:25:0 |
75.4:24.6:0 |
|
P2
|
5120 |
10700 |
2.08 |
50:50:0 |
48.8:51.2:0 |
|
P3
|
7400 |
14400 |
1.93 |
74:24:2 |
75:25:0 |
|
P4
|
11700 |
17900 |
1.53 |
72.5:22.5:5 |
70.2:25:4.8 |
|
P5
|
9630 |
16300 |
1.69 |
70:20:10 |
71.3:20:8.7 |
|
P6
|
8700 |
13600 |
1.56 |
60:20:20 |
65:22:13 |
|
P7
|
7000 |
11400 |
1.62 |
50:48:2 |
50:50:0 |
|
P8
|
6000 |
10000 |
1.66 |
50:45:5 |
50.3:45.5:4.2 |
|
P9
|
5000 |
10400 |
2.08 |
50:40:10 |
55:36.6:8.4 |
|
P10
|
5800 |
11400 |
1.96 |
50:30:20 |
53:31:16 |
Electrochemical characterization
The electrochemical behavior of the iridium monomers and all the copolymers were studied by cyclic voltammetric (CV) methods, and the relevant data are listed in Table 2. A quasi-reversible one-electron oxidation wave attributed to the oxidation of iridium(III) was detected at 0.47 and 0.32 V vs.Fc/Fc+ from Ir-complexes 3 and 4, respectively. However, these oxidation waves were elusive in the Ir-copolymers (P3–P10) possibly due to the low contents of Ir-complex 3. All copolymers (P1–P10) in this study exhibited oxidation potentials with the onsets lying in the range of 0.58–0.66 V vs.Fc/Fc+, which can be attributed to the oxidation of the copolymer backbones. The energies of the highest occupied molecular orbitals (HOMOs) in all copolymers were calculated relative to ferrocene (Fc) which has a value of 4.8 eV with respect to the vacuum level.21 The HOMO levels of copolymersP1–P10 (5.38–5.46 eV) were higher than that of PF (∼5.77 eV)22 because of the incorporation of BCB units. The energy band gaps of P1–P10 also increased slightly compared to that of PF, which can be attributed to deconjugation of the fluorenyl segments after introduction of BCB units. This result showed that the introduction of BCB units into the polymer main chains led to a rise in HOMO levels, which implied a much lower energy barrier for the hole injection. Similar behavior was also reported for polymerP(F-alt-Cz) with a similar backbone.23 The HOMO in combination with the optical band gaps derived from the absorption band edges were used to calculate the energies of the lowest unoccupied molecular orbital (LUMO) levels of the polymers. The HOMO and LUMO data are also demonstrated in Table 2.
Table 2 Electrochemical and thermal properties of Ir-complexes 3–4 and copolymersP1–P10
|
Copolymer or Ir-complex |
Eox
a/V |
HOMO b/eV |
LUMO c/eV |
E
g/eV |
T
d/°C |
|
Oxidation potential was adjusted by using ferrocene (E1/2 = 250 mV vs.Ag/AgNO3) as an internal reference. Conditions of cyclic voltammetric measurements: Ptworking electrode; Ag/AgNO3reference electrode. Scan rate: 100 mV s−1. Electrolyte: tetrabutylammonium hexafluorophosphate.
HOMO levels were calculated from CV potentials using ferrocene as a standard [HOMO = 4.8 + (Eox − EFc)].
LUMO levels were derived via eq. Eg = HOMO–LUMO, where Eg was obtained from the absorption spectra.
|
|
3
|
0.47 |
5.27 |
2.47 |
2.80 |
363 |
|
4
|
0.32 |
5.12 |
2.32 |
2.80 |
322 |
|
P1
|
0.60 |
5.40 |
2.48 |
2.92 |
409 |
|
P2
|
0.58 |
5.38 |
2.28 |
3.10 |
412 |
|
P3
|
0.62 |
5.42 |
2.30 |
2.92 |
401 |
|
P4
|
0.65 |
5.45 |
2.50 |
2.92 |
334 |
|
P5
|
0.63 |
5.43 |
2.51 |
2.92 |
320 |
|
P6
|
0.62 |
5.44 |
2.51 |
2.93 |
310 |
|
P7
|
0.61 |
5.41 |
2.31 |
3.10 |
385 |
|
P8
|
0.66 |
5.46 |
2.36 |
3.10 |
365 |
|
P9
|
0.63 |
5.43 |
2.33 |
3.10 |
343 |
|
P10
|
0.64 |
5.45 |
2.33 |
3.10 |
323 |
Thermal analysis
The thermal properties of the copolymers were investigated by differential scanning calorimeter (DSC) and thermal gravimetric analysis (TGA) under nitrogen. Thermal analysis data for the copolymers and iridium monomers are also illustrated in Table 2. All copolymers (P1–P10) and iridium complexes (3 and 4) exhibited good thermal stability with thermal decomposition temperatures (Td at 5% weight loss) ranging from 310 to 412 °C. In contrast, poly(9,9-diakylfluorene) was reported to have a thermal decomposition temperature at ∼390 °C under dry nitrogen.24 In our study, Td was observed to decrease as the feeding ratio of iridium units increased. No obvious phase transitions of all copolymers (P1–P10) were detected by DSC in the temperature range of 30–300 °C.
Optical properties
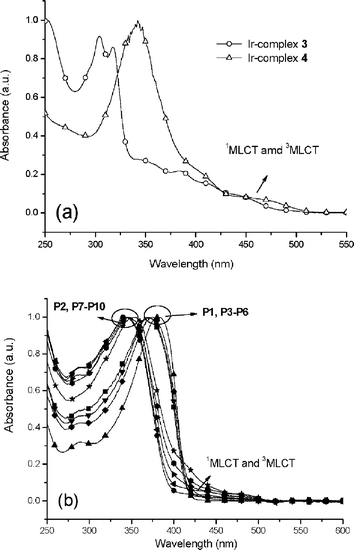
The UV-vis absorption spectra (in CH2Cl2 solutions) are shown in Fig. 1 and the photophysical data are illustrated in Table 3. In Fig. 1(a), iridium complexes 3 and 4 showed strong absorption bands at 273–370 nm attributed to the π–π* transition of benzimidazolyl ligands and weak bands at 400–500 nm attributed to the 1MLCT and 3MLCT transitions.19 In Fig. 1(b), the intense bands of all copolymers at ∼343–382 nm can be assigned to the π–π* transition of the polymer backbones and the benzimidazole ligands. Compared with P2 and P7–P10, the π–π* transition bands shifted to a shorter wavelength as the content of BCB in copolymersP1 and P3–P6 increased, respectively, indicating that the presence of 3,6-carbazole linkages interrupted the delocalization of π-electrons along the polymer backbones.25 For example, due to the higher content of BCB units in P2, the absorption peak in CH2Cl2 solution of P2 was blue shifted by 28 nm (371 to 343 nm) compared with that of P1. The weak MLCT bands at 400–500 nm were partially overlapped with the π–π* transition bands. The absorption spectra of copolymers were slightly red shifted in solid films due to the π–π stacking of molecules.
Table 3 Photophysical properties of Ir-complexes 3–4, copolymersP1–P8, and Ir-doped copolymers (P1–P2 doped with various mol% Ir-complex 4)
| |
In solution |
Solid film |
|
λ
max,abs/nm a |
λ
PL,max/nm |
E
T/eV |
ΦPL (%) |
λ
max,abs/nm f |
λ
PL,max/nmf |
E
T/eV |
ΦPL (%) h |
τ
|
|
Measured in CH2Cl2 solutions at 298 K at a concentration of 10−5 M.
Measured in toluene solutions at 298 K.
Triplet energy measured at 77 K in toluene solution.
Quantum yields were measured with respect to Ir(ppy)3 (ΦPL = 0.4 in toluene). The excitation wavelength was 400 nm for all Ir-complexes.
Measured in CH2Cl2 solutions in air relative to curmarin 1 (ΦPL = 0.99 in ethyl acetate) as a reference. The excitation wavelength was 350 nm for all copolymers.
Neat-film data were measured at 298 K.
Triplet energy measured at 77 K.
Neat-film data were measured at 298 K. PL quantum efficiencies in solid films were measured in an integrating sphere. The excitation wavelength was 350 nm for all copolymers.
Measured at 298 K. The excitation wavelength was 350 nm for all copolymers. The fluorescence lifetimes and phosphorescence lifetimes were monitored at 430 and 575 nm, respectively.
|
|
3
|
303, 316, 388, 416, 450 |
518 b |
2.40 |
30 d |
|
539 |
2.30 |
3.6 |
|
|
4
|
340, 400, 452, 472 |
566 b |
2.19 |
25 d |
|
578 |
2.14 |
2.5 |
|
|
P1
|
371 |
418 a |
2.38 c |
80 e |
372 |
446 |
2.30 g |
30 |
0.26 ns |
|
P2
|
343 |
409 a |
2.34 c |
75 e |
350 |
422 |
2.16 g |
25 |
0.38 ns (75.8%) |
| 1.69 ns (24.2%) |
|
P3
|
382 |
418 a |
|
56 e |
389 |
467 |
|
1.3 |
1.12 ns (47.5%) |
| 6.42 ns (40.4%) |
| 106.4 ns (12.1%) |
|
P4
|
368 |
418 a |
|
40 e |
372 |
583 |
|
0.6 |
0.29 μs |
|
P5
|
372 |
418 a |
|
24 e |
375 |
577 |
|
0.3 |
0.80 μs |
|
P6
|
343 |
419 a |
|
20 e |
376 |
582 |
|
0.5 |
0.85 μs |
|
P7
|
344 |
409 a |
|
53 e |
354 |
414 |
|
5.5 |
1.17 μs (78.7%) |
| 0.33 ns (21.3%) |
|
P8
|
347 |
410 a |
|
38 e |
352 |
568 |
|
2.0 |
1.12 μs |
|
P9
|
345 |
410 a |
|
26 e |
350 |
568 |
|
2.4 |
1.12 μs |
|
P10
|
345 |
410 a |
|
21 e |
350 |
568 |
|
2.1 |
1.12 μs |
|
P1 + 2 mol% 4 |
|
|
|
|
|
565 |
|
3.8 |
0.82 μs |
|
P1 + 5 mol% 4 |
|
|
|
|
|
565 |
|
2.1 |
0.75 μs |
|
P1 + 10 mol% 4 |
|
|
|
|
|
564 |
|
2.0 |
0.77 μs |
|
P1 + 20 mol% 4 |
|
|
|
|
|
564 |
|
2.1 |
0.69 μs |
|
P2 + 2 mol% 4 |
|
|
|
|
|
563 |
|
8.4 |
1.07 μs |
|
P2 + 5 mol% 4 |
|
|
|
|
|
563 |
|
6.9 |
1.15 μs |
|
P2 + 10 mol% 4 |
|
|
|
|
|
564 |
|
5.8 |
1.12 μs |
|
P2 + 20 mol% 4 |
|
|
|
|
|
564 |
|
5.0 |
0.97 μs |
The photoluminescence (PL) properties of Ir-complexes 3 and 4, copolymersP1–P10, and Ir-doped copolymers are given in Table 3. Representative PL spectra of the Ir-complexes 3 and 4 (in toluene) and selected copolymers (P5, P6, P9, and P10 in dichloromethane) are shown in Fig. 2(a) and 2(b), respectively. In Fig. 2(a), it is important to note that the PL emission of Ir-complex 4 is red shifted in comparison with that of (pbi)2Ir(acac), which was attributed to the extension of the ligand conjugation with fluorene units in Ir-complex 4. In Fig. 2(b), all copolymers (P1–P10) and Ir-doped copolymers (P1 and P2 with different mol% Ir-complex 4) emitted only characteristic violet-blue light in dilute dichloromethane solutions due to the π–π* transition of the polymers as excited at 350 nm. Apparently, energy transfer from the polymer backbone to the iridium unit was very inefficient in the solution either intramolecularly or intermolecularly. Fig. 2(c) and 2(d) showed the PL spectra of Ir-copolymers P3–P10 in solid films, and Fig. 2(e) and 2(f) were the PL spectra of Ir-doped copolymers in solid films. From P1 to P2, there were blue shifts of PL spectra in both CH2Cl2 solutions (9 nm) and solid films (24 nm), which was consistent with the trend observed in the absorption spectra. However, phosphorescence emission of Ir-copolymers (P3–P10) and Ir-doped copolymers from the iridium units was more obvious in solid films, especially for higher concentration (10 and 20 mol%) of iridium units, indicating the presence of energy transfer from π–π* transitions to MLCT bands. The efficiency of the energy transfer appeared to be higher as the BCB ratio in the polymer backbone increased, i.e.P8, P9, P10 and Ir-doped P2 were more efficient than P4, P5 and P6, and Ir-doped P1, respectively. Accordingly, the PL spectra of P4, P5, P6 and Ir-doped P1 exhibited more residual blue emissions. Besides the larger HOMO/LUMO gap, the larger triplet energy (ET), and thus less energy back transfer in P8, P9, P10 and Ir-doped P2 were believed to be also the cause of their higher phosphorescence efficiencies. This argument was supported by the longer phosphorescence lifetimes of P7–P10, and Ir-doped P2 than P3–P6 and Ir-doped P1. The former and the latter were measured to be 0.97–1.17 μs and 0.1–0.85 μs, respectively. Compared with the relative emission intensity of phosphorescence vs. fluorescence in the Ir-copolymers of Fig. 2(d), the energy transfer appeared to be more efficient in the Ir-doped copolymers of Fig. 2(f). This observation implied that the intermolecular energy transfer in Ir-doped copolymers occurred more readily than the main-chain intramolecular energy transfer in Ir-copolymers. In addition, the efficiency of the energy transfer increased as the iridium content increased in both Ir-doped copolymers and Ir-copolymers.
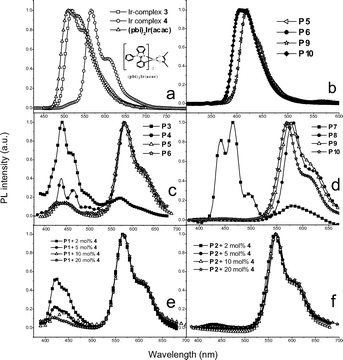 |
| | Fig. 2
PL spectra of (a) Ir-complexes 3, 4 and (pbi)2Ir(acac) in toluene solutions, (b) selected copolymers (P5, P6, P9, and P10) in CH2Cl2 solutions, (c) and (d) Ir-copolymers P3–P8 in solid films, and (e) and (f) Ir-doped copolymers in solid films. | |
The PL quantum yield (QY) values of the copolymers are also listed in Table 3. The phosphorescence QY values of Ir-complexes 3 and 4 were 30% and 25% in degassed toluene solutions, and 3.6% and 2.5% in solid films, respectively. Compared with the QY values of P1 and P2 in solid films (30% and 25%, respectively), Ir-copolymers P3–P10 and Ir-doped copolymersP1–P2 were found to have lower PL efficiencies (solid films) in the ranges of 0.3–5.5% and 1.3–8.4%, respectively.
Generally, the PL efficiencies decreased as the content of the iridium units increased. It can be rationalized by the greater tendency of triplet–triplet annihilation at higher iridium concentrations. The QY values of the Ir-copolymers and Ir-doped copolymers were higher as the BCB ratio in the polymer backbone increased, i.e., P8, P9, P10 and Ir-doped P2 were higher than P4, P5, P6 and Ir-doped P1, respectively. Quenching of the phosphorescence via energy back transfer of the phosphor excited state to the polymer triplet excited state has been well demonstrated for the polymer with lower triplet energy. For example, fluorene-alt-3,4-pyridinepolymers (PFPy) containing iridium complexes were reported to have lower phosphorescent QY than fluorene-alt-thiophenepolymers (PFT) because the lower triplet energy of PFPy (ET = 2.13 eV) than PFT (ET = 2.88 eV) led to more facile energy back transfer.10b As shown in Fig. 3, the solid film ET of P2 and P1 were measured to be 2.30 and 2.16 eV, respectively, and the ET of Ir-complex 4 was 2.14 eV (from phosphorescent emission wavelength). Fig. 4 shows a representative example illustrating the relative energy states for P1, P2 and Ir-complex 4 as well as the transition between different states. It was believed that the higher film QY values of the Ir-doped copolymers (P1 + 5, 10 and 20 mol% Ir-complex 4) compared to the Ir-copolymers (P4–P6) partially benefited from the less efficient energy back transfer in the former. The energy transfer efficiency depended on the distance, orientation, and overlapped area of absorption-PL spectra between the host and the guest. The slightly lower PL efficiencies of Ir-copolymers P3–P10 may be explained by the constrained orientation of iridium complexes covalently bonded to the polymer backbones as well as the larger π–π interactions induced by the polymer main chains, which diminished the mobility of the phosphorescent iridium moieties and thus hampered the energy transfer. Therefore, less flexibilities of polymer backbones result in less efficient energy transfers in Ir-copolymers.
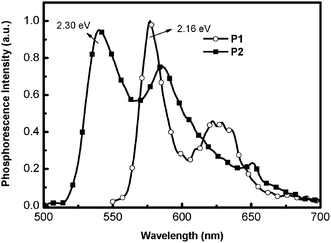 |
| | Fig. 3 Triplet energy level diagram of the P1 and P2 in solid films. | |
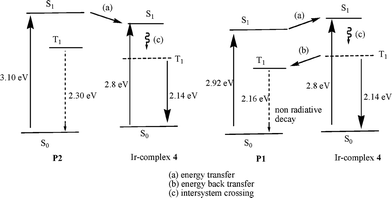 |
| | Fig. 4 Relative energy level diagram of the P1–P2 and Ir-complex 4. | |
Electroluminescent properties
PLED devices with a configuration of ITO/PEDOT: PSS (70 nm)/Ir-copolymers (P3–P10) or Ir-doped copolymers (P1–P2 doped with 2, 5, 10 or 20 mol% Ir-complex 4) (60–80 nm)/TPBI (40 nm)/LiF (1 nm)/Al (120 nm) were fabricated, where the vacuum deposited TPBI was used as an electron-transporting and hole-blocking layer. PLED devices without TPBI were excluded due to the extremely low efficiencies (reduced by at least one order of magnitude). The configuration of PLED devices and the chemical structures of materials used in this study are shown in Fig. 5(a) and the energy levels of PLED devices are shown in Fig. 5(b). The EL spectra and the EL performance data of all PLED devices are shown in Fig. 6 and Table 4, respectively.
Table 4
EL properties of PLED devices containing Ir-copolymers (P3–P10) and Ir-doped copolymers (P1–P2) doped with various mol% Ir-Complex 4a
| |
V
ON
|
L,max (at V) |
η
ext, max |
η
c, max |
λ
em,max at 10 V |
CIE at 10 V |
| V |
cd m−2
|
% |
cd A−1
|
nm |
x,y
|
|
V
on, turn-on voltage; L, luminance; V, voltage; ηext, external quantum efficiency; ηc, current efficiency.
|
|
P3
|
5.0 |
470 (18.0) |
0.59 |
0.61 |
436 |
0.16, 0.10 |
|
P4
|
4.5 |
1000 (18.0) |
0.82 |
1.53 |
428 |
0.27, 0.21 |
|
P5
|
4.5 |
1040 (17.5) |
1.15 |
2.87 |
580 |
0.40, 0.30 |
|
P6
|
4.0 |
3195 (13.5) |
0.94 |
2.47 |
580 |
0.50, 0.44 |
|
P7
|
4.5 |
1500 (15.0) |
0.89 |
1.55 |
422 |
0.25,0.19 |
|
P8
|
5.0 |
1960 (18.0) |
0.93 |
1.88 |
576 |
0.35, 0.32 |
|
P9
|
4.0 |
2300 (18.0) |
1.39 |
3.45 |
576 |
0.45, 0.38 |
|
P10
|
4.0 |
2176 (15.0) |
1.53 |
4.14 |
576 |
0.50, 0.45 |
|
P1 + 2 mol% 4 |
4.5 |
1810 (12.5) |
1.83 |
2.99 |
414 |
0.31, 0.26 |
|
P1 + 5 mol% 4 |
4.5 |
2030 (12.5) |
1.62 |
2.66 |
420 |
0.36, 0.25 |
|
P1 + 10 mol% 4 |
5.0 |
1980 (17.0) |
1.48 |
2.20 |
434 |
0.28, 0.21 |
|
P1 + 20 mol% 4 |
5.0 |
4950 (14.5) |
1.05 |
2.84 |
566 |
0.42, 0.41 |
|
P2 + 2 mol% 4 |
3.5 |
2710 (12.5) |
1.27 |
1.55 |
430 |
0.23, 0.16 |
|
P2 + 5 mol% 4 |
4.0 |
4870 (17.0) |
4.09 |
10.94 |
572 |
0.40, 0.35 |
|
P2 + 10 mol% 4 |
4.0 |
6150 (17.0) |
3.55 |
8.59 |
574 |
0.45, 0.44 |
|
P2 + 20 mol% 4 |
5.0 |
6820 (15.0) |
2.18 |
5.87 |
564 |
0.49, 0.51 |
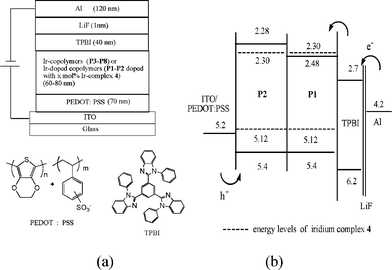 |
| | Fig. 5 (a) The configuration of PLED devices and the molecular structures of PEDOT and TPBI used in the devices and (b) relative energy levels of the compounds utilized in the PLED devices. | |
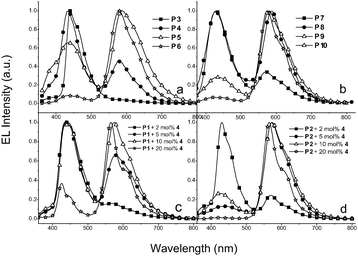 |
| | Fig. 6
EL spectra (at 10 V) of various PLED devices containing Ir-copolymers: (a) and (b), and Ir-doped copolymers: (c) and (d). | |
The EL performance parameters were in the order P7–P10 (ηext,max = 0.89–1.53%, and ηc,max = 1.55–4.14 cd A−1) > P3–P6 (ηext,max = 0.59–1.15%, and ηc,max = 0.61–2.87 cd A−1) and Ir-doped copolymerP2 (ηext,max = 1.27–4.09%, and ηc,max = 1.55–10.94 cd A−1) > P1 (ηext,max = 1.05–1.83%, and ηc,max = 2.20–2.99 cd A−1). The EL characteristic curves of current efficiency and luminance vs. current density of selected PLED devices are shown in Fig. 7.
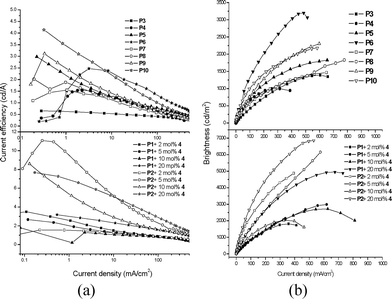 |
| | Fig. 7 The EL characteristic curves of PLED devices containing Ir-copolymers (P3–P10) and Ir-doped copolymers (a) current efficiency vs. current density plots and (b) luminance vs. current density plots. | |
Similar to the PL spectra in solid films, the EL spectra of PLED devices containing Ir-copolymers (P3–P10) (Fig. 6(a), and 6(b)) and Ir-doped copolymers (Fig. 6(c), and 6(d)) have both contributions from the polymer backbones and iridium moieties. However, the relative emission intensity of the iridium unit vs.polymer backbones in the EL spectra was different from that in the PL spectra. If the host does not have sufficiently high triplet energy, energy back transfer can occur readily, either intermolecularly26 or intramolecularly,27 which will counteract the contribution of phosphor molecule which has the theoretical maximum value of 75%. In addition to inefficient energy transfer from the host to the guest, energy back transfer from the guest to the host may be also the cause of the stronger blue emission in the EL spectra than that in the PL spectra. In this study, the ET of P2, P1, and Ir-complex 4 were 2.30, 2.16, and 2.14 eV, respectively (vide supra). Therefore, the energy back transfer from Ir-complex 4 to P1 had a greater tendency than that from 4 to P2 and the relative phosphorescence intensity of 4 to P1 is smaller than that of 4 to P2 in the PLED devices as shown in Fig. 6(c) and 6(d). Similarly, the energy back transfers of P3–P6 (Fig. 6(a)) were more facile than those of P7–P10 (Fig. 6(b)).
It was realized that the EL emission of the polymer backbone (with a shorter emission wavelength) increased relative to that of the iridium units (with a longer emission wavelength) as the operating voltage of PLED devices increased. Such an outcome may be attributed to the increased probability of energy back transfer because of the greater amount of triplet excitons generated at higher voltages. White light-emitting PLEDs may be possible form these materials possessing a dual emission (blue and yellow-orange) characteristic. Ir-copolymer P8 (with 5 mol% iridium units) and Ir-doped copolymerP2 (with 5 mol% Ir-complex 4) were therefore tested. The EL spectra and the CIE (Commossion Internationale d'Eclairage) coordinates of the two devices are shown in Fig. 8 and 9, respectively. At lower driving voltages, yellow-orange phosphorescence was prominent because of facile charge-trapping at the iridium unit. At higher driving voltages, the blue emission of the polymer backbone dominated over the phosphorescence because both singlet and triplet excitons of the polymer backbone increased significantly. The iridium phosphorescence was further suppressed at higher driving voltages because of higher concentrations of triplet excitons formed in the polymer backbones. The increased singlet excitons led to enhance the blue emission, while the increased triplet excitons suppressed the phosphorescence from the iridium units. The best EL properties of this study were found in PLED devices containing Ir-copolymers P8 (with ηext,max = 0.93%, ηc,max = 1.88 cd A−1, and Lmax = 1960 cd m−2) and P2 doped with 5 mol% Ir-complex 4 (with ηext,max = 4.09%, ηc,max = 10.94 cd A−1, and Lmax = 4870 cd m−2), respectively. The CIE coordinates, color-rendering index (CRI) and correlated color temperature (CCT) of the two PLED devices were (0.33, 0.30) at 13V, 74 and 5966 K for the former, and (0.35, 0.32) at 15 V, 82 and 6147 K for the latter, respectively.
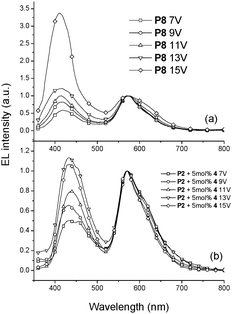 |
| | Fig. 8
EL spectra of PLED devices containing Ir-copolymers (with 5 mol% iridium units) (a) P8 and (b) P2 doped with 5 mol% Ir complex 4 at various voltages. | |
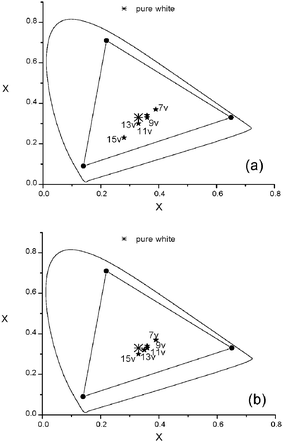 |
| | Fig. 9
CIE coordinates of various PLED devices containing (a) Ir-copolymers P8 and (b) Ir-doped P2 with 5 mol% Ir-complex 4 at low and high voltages (8–15 V). | |
Conclusions
In conclusion, a series of novel copolymers consisting of BCB segments and benzimidazole-based iridium units in the backbone by Suzuki coupling reaction were developed in this report. Incorporation of BCB units into the polymer backbones led to the rises in HOMO–LUMO bandgaps and the increases in triplet energy levels. As the content of BCB units in the copolymers increased, more efficient energy transfers were induced from the polymer backbones to the iridium units, and less efficient energy back transfers from the iridium units to the polymer backbones due to the enlarged triplet energy of the latter. Both Ir-copolymers (P3–P10) and Ir-doped copolymers (P1–P2 with Ir-complex 4) were used as the emitting layer of phosphorescent PLEDs. Copolymers with larger triplet energies of the polymer backbones had better performance due to more efficient suppression of energy back transfer. Less energy back transfer also led to better performance of the Ir-doped systems compared to the Ir-tethered systems. WOLED can be fairly achieved with the use of P8 and P2 doped with 5 mol% Ir-complex 4. The CIE coordinates, color-rendering index (CRI) and correlated color temperature (CCT) of the two PLED devices were (0.33, 0.30) at 13V, 74 and 5966 K from the former, and (0.35, 0.32) at 15 V, 82 and 6147 K from the latter, respectively.
Experimental
General methods
1H NMR spectra were recorded on a Bruker AMX400 spectrometer. FAB-mass spectra were collected on a JMS-700 double focusing mass spectrometer (JEOL, Tokyo, Japan) with a resolution of 3000 for low resolution and 8000 for high resolution (5% valley definition). The source accelerating voltage was operated at 10 kV with a Xe gun for FAB-mass spectra, using 3-nitrobenzyl alcohol as the matrix. The molecular weights of the polymers were determined by a Viscotek TriSEC GPC in THFsolvent. The number-average and weight-average molecular weights were estimated by using a calibration curve of polystyrene standards. Elemental analyses were performed on a Perkin-Elmer 2400 CHN analyzer. Cyclic voltammetry (CV) experiments were performed with a CHI-621B electrochemical analyzer and carried out at room temperature with a conventional three-electrode configuration consisting of a platinumworking electrode, an auxiliary electrode, and a nonaqueous Ag/AgNO3reference electrode. The solvent used in all CV experiments was CH2Cl2 and the supporting electrolyte was 0.1 M tetrabutylammoniumhexafluorophosphate (Bu4NPF6). The E1/2 values were determined to be as 1/2(Epa + Epc), where Epa and Epc were the anodic and cathodic peak potentials, respectively. All reported potentials were referenced to Fc+/Fc external standard (+0.250 V relative to the Ag/AgNO3electrode). Electronic absorption spectra were obtained on a Cary 50 ProbeUV-visible spectrometer. Emission spectra were recorded in deoxygenated solutions at 298 K by a JASCO FP-6500 fluorescence spectrometer. The emission spectra in solutions were collected on samples with O.D. ∼0.1 at the excitation wavelength. Emission maxima were reproducible within 2 nm. The fluorescence and phosphorescence solution quantum yields were calculated relative to coumarin 1 standard (Φem = 0.99 in ethyl acetate)28 and Ir(ppy)3 (Φem = 0.4 in toluene).29 Luminescence lifetimes were determined on an Edinburgh FL920 time-correlated pulsed single-photon-counting instrument. The solid film quantum yields were measured with an integrating sphere under an excitation wavelength of 350 nm on a quartz glass. Phosphorescence spectra of the compounds (in toluene solutions and solid films) were measured by a HORIBA Jobin-Yvon FluoroMax-P spectrometer at 77 K using a 10-ms delay time between the excitation with a microsecond flash lamp and the experiment. Luminescence quantum yields were taken by the average of three separate determinations and were reproducible within 10%. Differential scanning calorimetry (DSC) measurements were carried out from 30 to 300 °C using a Perkin-Elmer 7 series thermal analyzer at a heating and cooling rate of 10 °C min−1. Thermogravimetric analyses (TGA) were performed on a Perkin Elmer Pyris 1 TGA at a heating rate of 10 °C min−1 under nitrogen.
Device fabrication and measurement
Prepatterned ITO substrates with an effective individual device area of 3.14 mm2 were cleaned via repeated ultrasonic washing with detergent, deionized water, ethanol, and oxygen plasma treatment, subsequently. A layer of poly(ethylenedioxythiophene):poly(styrene-sulfonic acid) (PEDOT:PSS) (Baytron AI4083) with a thickness of 70 nm was spin-coated on the pre-cleaned ITO glass substrates as a hole injection layer and then baked at 100 °C in air for 1 h. After, the polymers were dissolved in chlorobenzene (concentration: 10 mg mL−1 for the polymers) and filtered with a 0.2 μm filter, and then a thin film of polymer was coated at a spin rate of 1800 rpm (revolution per min.). The film thickness of the polymer layer was around 40–60 nm, monitored by a surface profilometer dektak 3 (Veeco/ Sloan Instrument Inc.). Then, a layer of electron transporting 1,3,5-tris(N-phenylbenzimidazol-2-yl)benzene (TPBI) with a thickness of 40 nm was deposited under vacuum. Finally, a layer of LiF/Al (1 nm/ 120 nm) was thermally evaporated as a cathode in a vacuum chamber (under a pressure of less than 2.5 × 10−5 torr). I–V curves were measured on a Keithley 2400 Source Meter in ambient environment, and light intensities were measured with a Newport 1835 Optical Meter.
Materials
Chemicals and solvents were reagent grades and purchased from Aldrich, Acros, TCI, and Lancaster Chemical Co. Solvents were dried by standard procedures. All reactions and manipulations were carried out under N2 with the use of standard inert atmosphere and Schlenk techniques. All column chromatography was performed by using silica gel (230–400 mesh, Macherey-Nagel GmbH & Co.) as the stationary phase in a column with a length of 25–35 cm and a diameter of 2.5 cm.
Synthesis
2.4.1. 2-(4-Bromophenyl)-1-phenyl-1H-benzimidazole (1, pbi-Br).
N-Phenyl-o-phenylenediamine (1 eq.) and 4-bromobenzaldehyde (1 eq.) were dissolved in 50 mL of 2-methoxyethanol. The mixture was heated to reflux for 48 h under nitrogen. The volatiles were removed under vacuum and the resulting solid was extracted by dichloromethane. The organic extract was washed with brine solution, dried over anhydrous MgSO4, filtered and dried. The crude product was purified by column chromatography (silica gel) using a mixture of CH2Cl2 and hexanes (1:1 by volume) as the eluent to afford the pure compound as a white solid in 60% yield. 1H NMR (CDCl3, 400 MHz, ppm): δ 7.68 (d, J = 8.0 Hz, 1H), 7.35–7.29 (m, 4H), 7.24–7.20 (m, 1H), 7.15–7.02 (m, 7H). FABMS: m/z 348.9 (M) +. Anal. calcd. for C19H13BrN2: C, 65.35; H, 3.75; N, 8.02. Found: C, 65.22; H, 3.78; N, 8.01
2-(4-(9,9-Dihexyl-9H-fluoren-2-yl)phenyl)-1-phenyl-1H- benzimidazole (2 or pbiF).
To a mixture of toluene and aqueous solution of K2CO3 (1:1 v/v, 40 mL), compound 1 (1.39 g, 4 mmol) and 2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-9,9-dihexylfluorene (1.73 g, 4 mmol), and tetrakis(triphenylphosphine)palladium (Pd(PPh3)4) (100 mg, 0.04 mmol) were added to reflux for 24 h. After cooling, the reaction was quenched with water and the mixture was extracted with dicholormethane. The combined extracts were then washed with brine, dried over MgSO4, and evaporated to dryness. The crude product was isolated by column chromatography on a silica gel column using a mixture of CH2Cl2 and hexanes (1:4 by volume) as the eluent to afford the pure compound as a bright yellow powder in 40% yield. 1H NMR (CDCl3, 400 MHz, ppm): δ 8.19 (s, 1H), 7.92 (d, J = 8.0 Hz, 1H), 7.74 (d, J = 8.0 Hz, 2H), 7.70 (d, J = 8.4 Hz, 2H), 7.62 (d, J = 8.4 Hz, 2 H), 7.58–7.53 (m, 4H), 7.41∼7.27 (m, 8H), 2.01–1.97 (m, 4H), 1.13–1.03 (m, 12H), 0.75 (t, J = 7.5 Hz, 6H), 0.66–0.63 (m, 4H). FABMS: m/z 603.2 (M + H) + Anal. calcd. for C44H46N2: C, 87.66; H, 7.69; N, 4.65. Found: C, 87.42; H, 7.78; N, 4.41.
(pbi-Br)2Ir(acac) (3).
To a flask containing IrCl3·nH2O (176 mg, 0.5 mmol) and compound 1 (700 mg, 2.0 equiv), a mixture of 2-ethoxyethanol and water (3:1 v/v, 25 mL) was added. The mixture was then refluxed for 48 h and cooled to room temperature. After cooling, the reaction was quenched with water, extracted with dicholormethane, and dried under vacuum. The solid formed was collected by filtration and evaporation to give the crude product. The crude product of μ-chloro-bridged Ir(III) dimer was mixed with Na2CO3 (0.30 g, 3.0 mmol), 2,4-pentanedione (0.30 g, 3.0 mmol), and 2-methoxyethanol (20 mL) in a flask. The mixture was heated to reflux for 24 h. After cooling, the reaction was quenched with water and the mixture was extracted with dicholormethane. The combined extracts were then washed with brine, dried over MgSO4, and evaporated to dryness. The crude product was isolated by column chromatography on a silica gel column using a mixture of CH2Cl2 and hexanes (1:1 by volume) as the eluent to afford the pure compound as a yellow solid in 65% yield. 1H NMR (CDCl3, 400 MHz, ppm): δ 7.68−7.63 (m, 8H), 7.60−7.58 (m, 4H), 7.32−7.27 (m, 4H), 7.14−7.11 (m, 2H), 6.64 (dd, J = 8.0 Hz and J = 2.0 Hz, 2H), 6.49 (d, J = 2.0 Hz, 2H), 6.38 (d, J = 8.4 Hz, 2H), 5.25 (s, 1H), 1.84 (s, 6H). FABMS: m/z 986.0 (M)+. Anal. calcd. for C43H31Br2IrN4O2: C, 52.29; H, 3.16; N, 5.67. Found: C, 52.55; H, 3.26; N, 5.56.
(pbiF)2Ir(acac) (4).
Compound 4 was synthesized by the same procedure as illustrated for compound 3 except that compound 1 was used instead of compound 2. The product was isolated as an orange solid in 40% yield. 1H NMR (CDCl3, 400 MHz, ppm): δ 7.87 (s, 2H), 7.88 (dd, J = 7.2 Hz and 1.6 Hz, 2H), 7.69–7.55 (m, 10H), 7.50–7.48 (m, 2H), 7.46 (d, J = 8.0 Hz, 2H), 7.37–7.34 (m, 4H), 7.24–7.15 (m, 8H), 7.01 (s, 2H), 6.82 (s, 2H), 6.80 (dd, J = 8.0 Hz and 1.6 Hz, 2H), 6.60 (d, J = 8.0 Hz, 2H), 5.29 (s, 1H), 1.91 (s, 6H), 1.90–1.58 (m, 8H), 1.13–1.03 (m, 24H), 0.75 (t, J = 7.6 Hz, 12H), 0.66–0.63 (m, 8H). FABMS: m/z 1495.1 (M)+. Anal. calcd. for C93H97IrN4O2: C, 74.71; H, 6.54; N, 3.75. Found: C, 74.44; H, 6.38; N, 3.62.
1,4-Bis(9-octyl-9H-carbarzol-3yl)-2,5-dioctyloxy-benzene (5 or BCB).
To a mixture of toluene and aqueous solution of K2CO3 (1:1 v/v, 40 mL), 1,4-dibromo-2,5-bis(octyloxy)benzene (4.92 g, 10 mmol), 9-octyl-9H-carbazol-3-ylboronic acid (3.23 g, 10 mmol), and tetrakis(triphenylphosphine)palladium (120 mg) were added to reflux for 24 h. After cooling, the reaction was quenched with water and the mixture was extracted with dicholormethane. The combined extracts were then washed with brine, dried over MgSO4, and evaporated to dryness. The crude product was isolated by column chromatography on a silica gel column using a mixture of CH2Cl2 and hexanes (1:20 by volume) as the eluent. The compound 5 was obtained as a pale yellow oil in 75% yield. 1H NMR (CDCl3, 400 MHz, ppm): δ 8.36 (s, 2H), 8.11 (d, J = 8.0 Hz, 2H), 7.75 (d, J = 8.0 Hz, 2H), 7.47–7.39 (m, 6H), 7.24–7.19 (m, 2H), 7.15 (s, 2H), 4.31 (t, J = 7.2 Hz, 4H), 3.96 (t, J = 6.4 Hz, 4H), 1.92–1.88 (m, 4H), 1.70–1.66 (m, 4H), 1.41–1.16 (m, 40H), 0.86 (t, J = 7.2 Hz, 6H), 0.80 (t, J = 6.8 Hz, 6H). FABMS: m/z 889.4 (M)+.
1,4-Bis(6-bromo-9-octyl-9H-carbarzol-3yl)-2,5-dioctyloxy-benzene (6 or BCB-2Br).
Compound 5 (7.3 g, 8.2 mmol) was dissolved in 20 mL of dimethylformamide in a 250 mL one-necked flask. N-Bromosuccinimide (NBS) (3.06 g, 17.2 mmol) was dissolved in 20 mL of dimethylformamide and added into the reaction flask through a dropping funnel over a period of 30 min. The mixture was stirred at room temperature for 12 h. The reaction was quenched with water and extracted with dicholormethane. The combined extract was then washed with brine, dried over MgSO4, and evaporated to dryness. The crude product was isolated by column chromatography on a silica gel column using a mixture of CH2Cl2 and hexanes (1:20 by volume) as the eluent. Compound 6 was isolated as a white solid in 80% yield. 1H NMR (CDCl3, 400 MHz, ppm): δ 8.29 (d, J = 1.5 Hz, 2H), 8.20 (d, J = 1.8 Hz, 2H), 7.77 (dd, J = 8.4 and J = 1.5 Hz, 2H), 7.53 (dd, J = 8.7 and 1.8 Hz, 2H), 7.43 (d, J = 8.4 Hz, 2H), 7.28 (d, J = 8.7 Hz, 2H), 7.12 (s, 2H), 4.28 (t, J = 7.2 Hz, 4H), 3.97 (t, J = 6.3 Hz, 4H), 1.89–1.81 (m, 4H), 1.73–1.64 (m, 4H), 1.41–1.16 (m, 40H), 0.85 (t, J = 7.2 Hz, 6H), 0.79 (t, J = 6.8 Hz, 6H). FABMS: m/z 1044.4 (M)+.
General procedure for copolymerization by the Suzuki cross-coupling method
The following generalized procedure was used for the preparation of all copolymers. To a 50 mL, two-necked flask charged with a condenser, tricaprylymethylammonium chloride (Aliquat 336) (∼20 wt% based on the monomer), diboronate (compound 8, 1 equiv.), dibromide (compounds 3, 6, and 7, 1 equiv.), and Pd(PPh3)4 (25 mg, 0.005 equiv) were added. After the flask was evacuated and refilled with nitrogen for three times, toluene (1 mL) was added. Once all monomers were dissolved, an aqueous solution of K2CO3 (2 M, 1 mL) was added. The mixture was heated to 100 °C and stirred for 48 h under nitrogen. Phenyl-boronic acid (100 mg) was added and stirred at the same temperature for 12 h. Then, bromobenzene (0.5 mL) was added in the solution and heated for another 12 h. The mixture was cooled and poured into a mixture of methanol and water (100 mL, 2:1 v/v). The crude copolymer was filtered, washed with excess methanol, acetone, hexanes, and dried. The polymer was dissolved in CH2Cl2 and precipitated with methanol for two times. The product was further purified by flash chromatography using silica gel and a mixture of dichloromethane and THF (4:1) as the eluent. A general nomenclatures for the copolymers (P1–P10) with respect to the abbreviations of their monomers and their mol% were adopted. For example, PF74BCB24(pbi)Ir2 (P3) was synthesized from the composition of the following monomers: 2 mol% dibromo-substituted Ir-complex (3), 24 mol% BCB-2Br (6), and 74 mol% fluorenes (7 and 8, abbreviated as F).
PF75BCB25 (P1).
Following the general polymerization procedure, compound 6 (1.0 equiv), 2,7-dibromo-9,9-dioctyl-9H-fluorene (compound 7, 1.0 equiv) and 2,2′-(9,9-di-n-octyl-9H-fluorene-2,7-diyl)bis(4,4,5,5,-tetramethyl)-1,3,2-dioxaborolane (compound 8, 2.0 equiv) were used in this polymerization to acquire a bright green solid. Yield = 75%. 1H NMR (CDCl3, 400 MHz, ppm): δ 8.45–8.42 (d, carbazole ring), 7.85–7.78 (m, fluorene and carbazole ring), 7.72–7.65 (m, fluorene and carbazole ring), 7.50–7.40 (m, fluorene ring), 7.19 (s, -C6H2-), 4.39–4.31(m, N–CH2), 4.00–3.95 (m, O–CH2), 2.19–2.05 (fluorene-CH2), 2.01–1.94 (m, –C8H17), 1.70–1.53 (m, –C8H17), 1.46–1.10 (m, –C8H17), 0.86–0.60 (m, –C8H17). Anal. calcd. for (C29H40)75(C62H82N2O2)25: C, 87.16; H, 9.92; N, 1.36. Found: C, 86.86; H, 9.82; N, 1.24. GPC: Mw (weight-average molecular weight) = 33900 Da, DPI = 1.65.
PF50BCB50 (P2).
Following the general polymerization procedure, compound 6 (1.0 equiv) and compound 8 (1.0 equiv) were used in this polymerization to acquire a gray solid. Yield = 78%. 1H NMR (CDCl3, 400 MHz, ppm): δ 8.45–8.42 (d, carbazole ring), 7.85–7.78 (m, fluorene and carbazole ring), 7.72–7.65 (m, fluorene and carbazole ring), 7.50–7.40 (m, fluorene ring), 7.19 (s, -C6H2-), 4.39–4.31 (m, N–CH2), 4.00–3.95 (m, O–CH2), 2.19–2.05 (fluorene-CH2), 2.01–1.94 (m, –C8H17), 1.70–1.53 (m, –C8H17), 1.46–1.10 (m, –C8H17), 0.86–0.60 (m, –C8H17). Anal. calcd. for (C29H40)50(C62H82N2O2)50: C, 85.66; H, 9.64; N, 2.20. Found: C, 84.32; H, 9.32; N, 2.24. GPC: Mw = 10700 Da, DPI = 2.08.
PF74BCB24(pbi)Ir2 (P3).
Following the general polymerization procedure, compound 3 (1.0 equiv), compound 6 (12.0 equiv), compound 7 (12.0 equiv), and compound 8 (25.0 equiv) were used in this polymerization to acquire a yellow-green solid. Yield = 72%. Anal. calcd. for (C29H40)74(C62H82N2O2)24(C43H31IrN4O2)2: C, 86.40; H, 9.73; N, 1.52. Found: C, 85.44; H, 9.32; N, 1.43. GPC: Mw = 14400 Da, DPI = 1.93.
PF72.5BCB22.5(pbi)Ir5 (P4).
Following the general polymerization procedure, compound 3 (5.0 equiv), compound 6 (22.5 equiv), compound 7 (22.5 equiv), and compound 8 (50.0 equiv) were used in this polymerization to acquire a yellow solid. Yield = 75%. Anal. calcd. for (C29H40)72.5(C62H82N2O2)22.5(C43H31IrN4O2)5: C, 85.29; H, 9.45; N, 1.74. Found: C, 84.82; H, 9.22; N, 1.44. GPC: Mw = 17900 Da, DPI = 1.53.
PF70BCB20(pbi)Ir10 (P5).
Following the general polymerization procedure, compound 3 (1.0 equiv), compound 6 (2.0 equiv), compound 7 (2.0 equiv), and compound 8 (5.0 equiv) were used in this polymerization to acquire a yellow solid. Yield = 72%. 1H NMR (CDCl3, 400 MHz, ppm): δ 8.45–8.42 (d, carbazole ring), 7.80–7.65 (m, fluorene and carbazole ring), 7.54–7.38 (m, fluorene ring), 7.19–7.16 (m, –C6H2-), 7.04–7.03 (m, benzimidazole ring), 6.75–6.70 (m, benzimidazole ring), 6.44–6.43 (m, benzimidazole ring), 4.39–4.31(m, N–CH2), 4.00–3.95 (m, O–CH2), 2.19–2.05 (fluorene-CH2), 2.01–1.94 (m, –C8H17), 1.70–1.53 (m, –C8H17), 1.46–1.10 (m, –C8H17), 0.86–0.60 (m, –C8H17). Anal. calcd. for (C29H40)70(C62H82N2O2)20(C43H31IrN4O2)10: C, 83.49; H, 8.99; N, 2.11. Found: C, 82.87; H, 9.12; N, 2.20. GPC: Mw = 16300 Da, DPI = 1.69.
PF60BCB20(pbi)Ir20 (P6).
Following the general polymerization procedure, compound 3 (2.0 equiv), compound 6 (2.0 equiv), compound 7 (1.0 equiv), and compound 8 (5.0 equiv) were used in this polymerization to acquire an orange solid. Yield = 72%. 1H NMR (CDCl3, 400 MHz, ppm): δ 8.45–8.42 (d, carbazole ring), 7.80–7.65 (m, fluorene and carbazole ring), 7.54–7.38 (m, fluorene ring), 7.19–7.16 (m, –C6H2-), 7.04–7.03 (m, benzimidazole ring), 6.75–6.70 (m, benzimidazole ring), 6.44–6.43 (m, benzimidazole ring), 4.39–4.31(m, N–CH2), 4.00–3.95 (m, O–CH2), 2.19–2.05 (fluorene-CH2), 2.01–1.94 (m, –C8H17), 1.70–1.53 (m, –C8H17), 1.46–1.10 (m, –C8H17), 0.86–0.60 (m, –C8H17). Anal. calcd. for (C29H40)60(C62H82N2O2)20(C43H31IrN4O2)20: C, 80.04; H, 8.15; N, 2.92. Found: C, 78.24; H, 9.75; N, 2.42. GPC: Mw = 13600 Da, DPI = 1.56.
PF50BCB48(pbi)Ir2 (P7).
Following the general polymerization procedure, compound 3 (1.0 equiv), compound 6 (24.0 equiv), and compound 8 (25.0 equiv) were used in this polymerization to acquire the product as a yellow solid. Yield = 85%. Anal. calcd. for (C29H40)50(C62H82N2O2)48(C43H31IrN4O2)2: C, 85.10; H, 9.49; N, 2.29. Found: C, 84.41; H, 9.24; N, 1.79. GPC: Mw = 11400 Da, DPI = 1.62.
PF50BCB45(pbi)Ir5 (P8).
Following the general polymerization procedure, compound 3 (1.0 equiv), compound 6 (9.0 equiv), and compound 8 (25.0 equiv) were used in this polymerization to acquire a yellow solid. Yield = 76%. Anal. calcd. for (C29H40)50(C62H82N2O2)45(C43H31IrN4O2)5: C, 84.26; H, 9.28; N, 2.43. Found: C, 84.41; H, 9.24; N, 2.39. GPC: Mw = 10000 Da, DPI = 1.66.
PF50BCB40(pbi)Ir10 (P9).
Following the general polymerization procedure, compound 3 (1.0 equiv), compound 6 (4.0 equiv), and compound 8 (5.0 equiv) were used in this polymerization to acquire a brown solid. Yield = 54%. 1H NMR (CDCl3, 400 MHz, ppm): δ 8.45–8.42 (d, carbazole ring), 7.80–7.65 (m, fluorene or carbazole ring), 7.54–7.38 (m, fluorene ring), 7.19–7.16 (m, –C6H2–), 7.04–7.03 (m, benzimidazole ring), 6.75–6.70 (m, benzimidazole ring), 6.44–6.43 (m, benzimidazole ring), 4.39–4.31 (m, N–CH2), 4.00–3.95 (m, O–CH2), 2.19–2.05 (fluorene-CH2), 2.01–1.94 (m, –C8H17), 1.70–1.53 (m, –C8H17), 1.46–1.10 (m, –C8H17), 0.86–0.60 (m, –C8H17). Anal. calcd. for (C29H40)50(C62H82N2O2)40(C43H31IrN4O2)10: C, 82.85; H, 8.91; N, 2.66. Found: C, 82.41; H, 8.64; N, 2.38. GPC: Mw = 10400 Da, DPI of 2.08.
PF50BCB30(pbi)Ir20 (P10).
Following the general polymerization procedure, compound 3 (2.0 equiv), compound 6 (3.0 equiv), and compound 8 (5.0 equiv) were used in this polymerization to acquire a brown solid. Yield = 54%. 1H NMR (CDCl3, 400 MHz, ppm): δ 8.45–8.42 (d, carbazole ring), 7.80–7.65 (m, fluorene or carbazole ring), 7.54–7.38 (m, fluorene ring), 7.19–7.16 (m, –C6H2-), 7.04–7.03 (m, benzimidazole ring), 6.75–6.70 (m, benzimidazole ring), 6.44–6.43 (m, benzimidazole ring), 4.39–4.31 (m, N–CH2), 4.00–3.95 (m, O–CH2), 2.19–2.05 (fluorene-CH2), 2.01–1.94 (m, –C8H17), 1.70–1.53 (m, –C8H17), 1.46–1.10 (m, –C8H17), 0.86∼0.60 (m, –C8H17). Anal. calcd. for (C29H40)50(C62H82N2O2)30(C43H31IrN4O2)20: C, 79.99 H, 8.18; N, 3.13. Found: C, 77.41; H, 7.14; N, 2.88. GPC: Mw = 11400 Da, DPI of 1.96.
Acknowledgements
The authors express their sincere thanks to the Academia Sinica, National Chiao Tung University, and the National Science Council for supporting this work.
Notes and references
- C. W. Tang and S. A. Van Slyke, Appl. Phys. Lett., 1987, 51, 913 CrossRef CAS.
- J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burn and A. B. Holmes, Nature, 1990, 347, 539 CrossRef CAS.
-
(a) M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151 CrossRef CAS;
(b) M. A. Baldo, D. F. O'Brien, M. E. Thompson and S. R. Forrest, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 14422 CrossRef CAS.
-
(a) H. Z. Xie, M. W. Liu, O. Y. Wang, X. H. Zhang, C. S. Lee, L. S. Hung, S. T. Lee, P. F. Teng, H. L. Kwong, H. Zheng and C. Che, Adv. Mater., 2001, 13, 1245 CrossRef CAS;
(b) V. V. Grushin, N. Herron, D. D. LeCloux, W. J. Marshall, V. A. Petrov and Y. Wang, Chem. Commun., 2001, 1494 RSC;
(c) J. Ostrowski, M. R. Robinson, A. J. Heeger and G. C. Bazan, Chem. Commun., 2002, 784 RSC;
(d) J.-P. Duan, P.-P. Sun and C.-H. Cheng, Adv. Mater., 2003, 15, 224 CrossRef CAS;
(e) Y.-J. Su, H.-L. Huang, C.-L. Li, C.-H. Chien, Y.-T. Tao, P.-T. Chou, S. Datta and R.-S. Liu, Adv. Mater., 2003, 15, 884 CrossRef CAS;
(f) A. B. Tamayo, B. D. Alleyne, P. I. Djurovich, S. Lamansky, I. Tsyba, N. N. Ho, R. Bau and M. E. Thompson, J. Am. Chem. Soc., 2003, 125, 7377 CrossRef CAS.
-
(a) W. Lu, B.-X. Mi, M. C. W. Chan, Z. Hui, N. Zhu, S.-T. Lee and C.-M. Che, Chem. Commun., 2002, 206 RSC;
(b) B. W. D'Andrade, J. Brooks, V. Adamovich, M. E. Thompson and S. R. Forrest, Adv. Mater., 2002, 14, 1032 CrossRef.
-
(a) Y. Ma, H. Zhang, J. Shen and C.-M. Che, Synth. Met., 1998, 94, 245 CrossRef CAS;
(b) J. Lu, Y. Tao, Y. Chi and Y.-Y. Tung, Synth. Met., 2005, 155, 56 CrossRef CAS;
(c) Y.-H. Niu, Y.-L. Tung, Y. Chi, C.-F. Shu, J. H. Kim, B. Chen, J. Luo, A. J. Carty and A. K.-Y. Jen, Chem. Mater., 2005, 17, 3532 CrossRef CAS;
(d) C.-H. Chien, P.-I. Shih, F.-I. Wu, C. F. Shu and Y. Chi, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2073 CrossRef CAS.
-
(a) H. Xia, C. Zhang, X. Liu, S. Qiu, P. Lu, F. Shen, J. Zhang and Y. Ma, J. Phys. Chem. B, 2004, 108, 3185 CrossRef CAS;
(b) F. G. Gao and A. J. Bard, J. Am. Chem. Soc., 2000, 122, 7426 CrossRef CAS.
-
(a) Y. Y. Chen, Y. T. Tao and H. C. Lin, Macromolecules, 2006, 39, 8559 CrossRef CAS;
(b) Y. Y. Chen and H. C. Lin, Polymer, 2007, 48, 5268 CrossRef CAS;
(c) Y. Y. Chen and H. C. Lin, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3243 CrossRef CAS;
(d) K. L. Paik, N. S. Baek and H. K. Kim, Macromolecules, 2002, 35, 6782 CrossRef CAS;
(e) X. Gong, D. Moses, A. J. Heeger and S. Xiao, J. Phys. Chem. B, 2004, 108, 8601 CrossRef CAS;
(f) S.-C. Lo, G. J. Richard, J. P. J. Markham, E. B. Namdas, S. Sharma, P. L. Burn and I. D. W. Samuel, Adv. Funct. Mater., 2005, 15, 1451 CrossRef CAS;
(g) S.-C. Lo, N. A. H. Male, J. P. J. Markham, S. W. Magennis, P. L. Burn and I. D. W. Samuel, Adv. Mater., 2002, 14, 975 CrossRef CAS.
-
(a) B. W. D. Andrade, M. E. Thompson and S. R. Forrest, Adv. Mater., 2002, 14, 147 CrossRef CAS;
(b) J. Feng, F. Li, W. Gao, S. Liu, Y. Liu and Y. Wang, Appl. Phys. Lett., 2001, 78, 3947 CrossRef CAS.
-
(a) J. R. Carlise, X. Y. Wang and M. Weck, Macromolecules, 2005, 38, 9000 CrossRef CAS;
(b) C. L. Schulz, X. Chen, S.-A. Chen and S. Holdcroft, Macromolecules, 2006, 39, 9157 CrossRef CAS;
(c) N. R. Evans, L. S. Devi, C. S. K. Mak, S. E. Watkins, S. I. Pascu, A. Köhler, R. H. Friend, C. K. Willans and A. B. Holms, J. Am. Chem. Soc., 2006, 128, 6647 CrossRef CAS.
-
(a) X. Gong, M. R. Robinson, J. C. Ostrowski, D. Moses, G. C. Bazan and A. J. Hegger, Adv. Mater., 2002, 14, 581 CrossRef CAS;
(b) Y. Kawamura, S. Yanagida and S. R. Forrest, J. Appl. Phys., 2002, 92, 87 CrossRef CAS;
(c) K.-M. Yeh, C.-C. Lee and Y. Chen, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5180 CrossRef CAS;
(d) C.-F. Liao, B.-Y. Hsieh and Y. Chen, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 149 CrossRef CAS.
-
(a) H. Zhen, C. Luo, W. Yang, W. Song, B. Du, J. Jiang, C. Jiang, Y. Zhang and Y. Cao, Macromolecules, 2006, 39, 1693 CrossRef CAS;
(b) K. Zhang, Z. Chen, Y. Zou, C. Yang, J. Qin and Y. Cao, Organometallics, 2007, 26, 3699 CrossRef CAS.
-
(a) X. W. Chen, J. L. Liao, Y. M. Liang, M. O. Ahmed, H. E. Tseng and S.-A. Chen, J. Am. Chem. Soc., 2003, 125, 636 CrossRef CAS;
(b) J. Jiang, C. Jiang, W. Yang, H. Zhen, F. Huang and Y. Cao, Macromolecules, 2005, 38, 4072 CrossRef CAS.
-
(a) P.-I. Lee and S. L.-C. Hsu, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1492 CrossRef CAS;
(b) K. Zhang, Z. Chen, C. Yang, Y. Zou, S. Gong, Y. Tao, J. Qin and Y. Cao, J. Mater. Chem., 2008, 18, 3366 RSC.
-
(a) Q. Pei and Y. Yang, J. Am. Chem. Soc., 1996, 118, 7416 CrossRef CAS;
(b) J. Jacob, J. Zhang, A. C. Grimsdale, K. Müllen, M. Gaal and E. J. W. List, Macromolecules, 2003, 36, 8240 CrossRef CAS;
(c) H.-J. Cho, B.-J. Jung, N. S. Cho, J. Lee and H.-K. Shim, Macromolecules, 2003, 36, 6704 CrossRef CAS;
(d) B. J. Jung, J.-I. Lee, H. Y. Chu, L.-M. Do and H.-K. Shim, Macromolecules, 2002, 35, 2282 CrossRef CAS.
- J. X. Jiang, Y. H. Xu, W. Yang, R. Guan, Z. Q. Lin, H. Y. Zhen and Y. Cao, Adv. Mater., 2006, 18, 1769 CrossRef CAS.
- F.-I. Wu, X.-H. Yang, D. Neher, R. Dodda, Y.-H. Tseng and C. F. Shu, Adv. Funct. Mater., 2007, 17, 1085 CrossRef CAS.
- P.-I. Lee, S. L.-C. Hsu and J.-F. Lee, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 464 CrossRef CAS.
- W.-S. Huang, J. T. Lin, C.-H. Chien, Y.-T. Tao, S.-S. Sun and Y.-S. Wen, Chem. Mater., 2004, 16, 2480 CrossRef CAS.
-
(a) W.-S. Huang, J. T. Lin and H.-C. Lin, Org. Electron., 2008, 9, 557 CrossRef CAS;
(b) W.-S. Huang, C.-W. Lin, J. T. Lin, J.-H. Huang, C.-W. Chu, Y.-S. Wu and H.-C. Lin, Org. Electron., 2009, 10, 594 CrossRef CAS.
- J. Pommerehne, H. Vestweber, W. Guss, R. F. Mahrt, H. Bässler, M. Porsch and J. Daub, Adv. Mater., 1995, 7, 551 CrossRef CAS.
- H. Y. Zhen, C. Y. Jiang, W. Yang, J. X. Jiang, F. Huang and Y. Cao, Chem.–Eur. J., 2005, 11, 5007 CrossRef CAS.
- Y. Li, J. F. Ding, M. Day, Y. Tao, J. P. Lu and M. D'iorio, Chem. Mater., 2004, 16, 2165 CrossRef CAS.
- B. Liu, W.-L. Yu, Y.-H. Lai and W. Huang, Chem. Mater., 2001, 13, 1984 CrossRef CAS.
- C. Xia and R. C. Advincula, Macromolecules, 2001, 34, 5854 CrossRef CAS.
-
(a) C. Adachi, R. C. Kwong, P. Djurovich, V. Adamovich, M. A. Baldo, M. E. Thompson and S. R. Forrest, Appl. Phys. Lett., 2001, 79, 2082 CrossRef CAS;
(b) M. Sudhakar, P. I. Djurovich, T. E. Hogen-Esch and M. E. Thompson, J. Am. Chem. Soc., 2003, 125, 7796 CrossRef CAS.
- Y. You, S. H. Kim, H. K. Jung and S. Y. Park, Macromolecules, 2006, 39, 349 CrossRef CAS.
- G. Jones II, W. R. Jackson, C. Y. Choi and W. R. Bwrgmark, J. Phys. Chem., 1985, 89, 294 CrossRef.
- A. Tsuboyama, H. Iwawaki, M. Furugori, T. Mukaide, J. Kamatani, S. Igawa, T. Moriyama, S. Miura, T. Takiguchi, S. Okada, M. Hoshino and K. Ueno, J. Am. Chem. Soc., 2003, 125, 12971 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.