DOI:
10.1039/B9PY00339H
(Paper)
Polym. Chem., 2010,
1, 485-493
Received
5th November 2009
, Accepted 13th December 2009
First published on 11th January 2010
Abstract
A novel spiro(fluorene-9,9′-xanthene) skeleton bis(ether amine) monomer, 2′,7′-bis(4-aminophenoxy)-spiro(fluorene-9,9′-xanthene), was prepared through a simple acid-catalyzed condensation reaction of 9-fluorenone with resorcinol to form the spiro framework through an sp3carbon atom. Subsequent nucleophilic substitution reaction of spiro[fluorene-9,9′-(2′,7′-dihydroxyxanthene)] with 1-fluoro-4-nitro-benzene in the presence of potassium carbonate in N,N-dimethylacetamide, followed by catalyticreduction with hydrazine and Pd/C in ethanol. A series of new polyimides were synthesized from the diamine with various commercially available aromatic tetracarboxylic dianhydrides via a conventional two-stage process with the thermal or chemical imidization of the poly(amic acid) precursors. Most of the polyimides obtained from both routes were soluble in many organic solvents such as N-methyl-2-pyrrolidone, N,N-dimethylacetamide and m-cresol. All the polyimides could afford transparent, flexible, and strong films with low moisture absorptions of 0.36–0.73% and low dielectric constants of 2.65–3.12 at 1 kHz. Thin films of these polyimides showed an UV-vis absorption cutoff wavelength at 352–409 nm, and those of polyimides from 4,4′-oxydiphthalic dianhydride and 2,2-bis(3,4-dicarboxyphenyl) hexafluoropropane dianhydride (6FDA) were essentially colorless. The polyimides exhibited excellent thermal stability, with decomposition temperatures (at 10% weight loss) above 510 °C in both air and nitrogen atmospheres and glass transition temperatures (Tg) in the range 296–346 °C.
1. Introduction
Aromatic polyimides are well known as high performance polymeric materials because of their good chemical resistance, high mechanical strength, and excellent thermal stability.1–3 Incorporation of specific functionality into the polyimide backbones leads to various advanced functional materials that exhibit certain advantageous properties, such as electrochromic, gas separation, charge-transporting, nonlinear optical, highly refractive, and photosensitive properties.4–9 However, their applicability has been limited because aromatic polyimides are normally insoluble and fusible in the fully imidized form because of their rigid-chain characteristics, leading to processing difficulties. Thus, polyimide processing is generally carried out with a poly(amic acid) intermediate and then is converted to a polyimide via rigorous thermal treatment. Problems often arise because the poly(amic acid)s are thermally and hydrolytically unstable. To overcome these problems, polymer structure modification becomes necessary. Therefore, several modification of the chemical structure have been made to enhance their processability and solubility while other advantageous polymer properties are retained either by introducing flexible linkages,10–16 bulky substituents,17–20 noncoplannar structures,21–23 and spiroskeletons24,25 into the polymer backbone.
It has been demonstrated that incorporating a spiroskeletons linkage into the structure of small molecules, as well as polymeric materials, leads to amorphous materials with an improvement in both solubility and thermal stability.26–28 In the spiro-segment, the rings of the connected spiroskeletons are orthogonally arranged and connected via a common tetra-coordinated carbon,29,30 and the polymer chains were twisted at an angle of 90° at each spiro-center. This structural feature was predicted to restrict the close packing of the polymer chains, thereby reducing the probability of interchain interactions, resulting in higher polymer solubility. Moreover, for the spiro-annulated segment, the rigidity of the polymer backbone would be preserved. Such spiro structures have also been applied to polymeric materials such as polyfluorenes,31–33 polyquinolines,34 and polyimides35–40 to reduce crystallization tendency and to enhance their solubility, glass-transition temperature (Tg), and thermal stability. More importantly, the fluorene containing polymers have been proven to exhibit high refractive indices due to their high aromatic contents, so some of the fluorene-containing polymers have been successfully utilized to make optical lenses.41 On the other hand, the incorporation of bulky fluorenegroups, which sterically hinder the intermolecular interaction of the PI chains, reduces the packing density, thus increases the transparency of the PIs.42,43
In this study, therefore, we attempted to synthesize a novel diamine monomer by combining spiro fluorene and xanthene moieties to afford good solubility and good thermal properties, as well as good dielectric and optical properties. A novel monomer, 2′,7′-bis(4-aminophenoxy)-spiro(fluorene-9,9′-xanthene) (3), was synthesized. This monomer was used to prepare polyimides with various commercially available aromatic tetracarboxylic dianhydrides via a conventional two-step process of poly(amic acid) preparation, which was followed by the thermal or chemical imidization. The polyimides synthesized were characterized by Fourier transform infrared (FTIR), NMR, gel permeation chromatography (GPC), differential scanning calorimetry (DSC), and thermogravimetric analysis (TGA), and their solubility, water absorption and dielectric constant were also measured.
2. Experimental
2.1 Materials
9-Fluorenone (Fluka), 1-fluoro-4-nitrobenzene (TCI, Japan), resorcinol (TCI), potassium carbonate (K2CO3) (Fluka), 10% palladium on charcoal (Pd/C) (Fluka), and hydrazine monohydrate (Acros) were used as received. Commercially available aromatic tetracarboxylic dianhydrides such as pyromellitic dianhydride (PMDA) (4a) (Aldrich) and 3,3′,4,4′-benzophenonetetracarboxylic dianhydride (BTDA) (4c) (Aldrich) were purified by recrystallization from acetic anhydride. 3,3′,4,4′-Biphenyltetracarboxylic dianhydride (BPDA) (4b) (Oxychem), and 4,4′-oxydiphthalic dianhydride (ODPA) (4d) (Oxychem) and 2,2-bis(3,4-dicarboxyphenyl)hexafluoropropane dianhydride (6FDA) (4e) (Hoechst Celanese) were heated at 250 °C under vacuum for 3 h prior to use. N,N-Dimethylacetamide (DMAc) was purified by distillation under reduced pressure over calcium hydride and stored over 4 Å molecular sieves.
2.2 Characterizations
The inherent viscosities of the polymers were measured with an Ubbelohde viscometer at 30 °C. Weight-average molecular weights (Mw) and number-average molecular weights (Mn) were obtained viagel permeation chromatography (GPC) on the basis of polystyrene calibration using Waters 2410 as an apparatus and tetrahydrofuran (THF) as the eluent. FT-IR spectra (KBr) were recorded on a Nicolet NEXUS670 fourier transform infrared spectrometer. 1H NMR and 13C NMR spectra were measured on a JEOL EX-300 spectrometer using tetramethylsilane as the internal reference. Elemental analyses were determined by a Perkin–Elmer model 2400 CHN analyzer. Testing by differential scanning calorimetry (DSC) was performed on a Perkin–Elmer differential scanning calorimeterDSC 7 or Pyris 1 DSC at a scanning rate of 20 °C min−1 in flowing nitrogen (30 cm3 min−1), and glass transition temperatures (Tg) were read at the DSC curves at the same time. Thermogravimetric analysis (TGA) was conducted with a TA Instruments TGA 2050, and experiments were carried out on approximately 10 mg of sample in flowing air (flowing rate = 100 cm3 min−1) at a heating rate of 20 °C min−1. The ultraviolet-visible (UV-vis) spectra were recorded on a Hitachi U-3210 spectrophotometer. Dielectric property of the polymer films was tested by the parallel-plate capacitor method using a HP-4194A Impedance/Gain Phase Analyzer. Goldelectrodes were vacuum deposited on both surfaces of dried film. Experiments were performed at 25 °C in a dry chamber. Elemental analyses were performed on a Yanaco MT-6 CHN recorder elemental analysis instrument. Wide-angle X-ray diffraction measurements were performed at room temperature (about 25 °C) on a Siemens Kristalloflex D5000 X-ray diffractometer, using nickel-filtered Cu-Kα, radiation (λ = 1.5418 Å, operating at 40 kV and 30 mA). Tensile properties were determined from stress-strain curves with a Toyo Instron UTM-III-500 with a load cell of 10 kg at a drawing speed of 5 cm min−1. Measurement were performed at 28 °C with film specimens (about 0.1 mm thick,1.0 cm wide and 5-cm long) and average of at least five individual determinations was used. The equilibrium water absorption was determined by the weighing of the changes in vacuum-dried film specimens before and after immersion in deionized water at 25 °C for 3 days.
2.3 Monomer synthesis
2.3.1 Synthesis of spiro[fluorene-9,9′-(2′,7′-dihydroxyxanthene)](1).
A mixture of 9-fluorenone (3.60 g, 20 mmol), rensorcinol (8.80 g, 80 mmol), and ZnCl2 (1.11 g, 9.01 mmol) was stirred and heated at 140 °C for 3 h. The melt was then heated with concentrated HCl (aqueous, 50 mL), under refux for another 2 h. The reaction mixture was poured into water (500 mL). The precipitate was washed with water, dried, and purified by column chromatography (EtOAc–hexane, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) to yield 1(4.88 g, 67.0%); mp = 266–268 °C (onset to peak top temperature), by differential scanning calorimetry (DSC) at a scan rate of 2 °C min−1.
:4) to yield 1(4.88 g, 67.0%); mp = 266–268 °C (onset to peak top temperature), by differential scanning calorimetry (DSC) at a scan rate of 2 °C min−1.
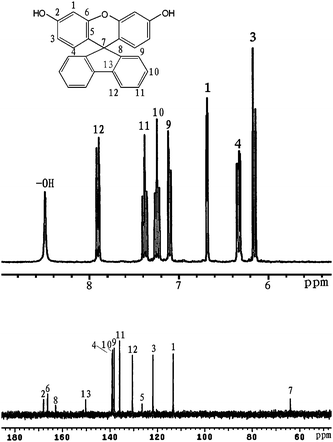
1H NMR (acetone-d6): δ6.13 (d, J = 8.7 Hz, 2H, H3), 6.31 (dd, J = 6.0, 2.7 Hz, 2H, H4), 6.67 (d, J = 2.4 Hz, 2H, H1), 7.08 (d, J = 7.8 Hz,2H, H9), 7.21 (dd, J = 7.2, 7.6 Hz, 2H, H10), 7.41 (dd, J = 7.2, 7.6 Hz, 2H, H11), 7.88 (d, J = 7.8 Hz, 2H, H12), 8.49(s, 2H,–OH). 13C NMR (acetone-d6): 63.4 (C7), 113.3 (C1), 121.8 (C3), 126.4 (C5), 130.5 (C12), 135.9 (C11), 138.2 (C9), 138.8 (C10), 139.1 (C4), 150.1 (C13), 162.7 (C8), 166.1 (C6), 167.8 (C2). FT-IR (KBr): 3504–3404 cm−1 (O–H), 3063–3033 cm−1(C–H), 1168 and 1260 cm−1(C–O). EI-MS (m/z):[M]+ calcd for C25H16O3, 364; Found 364. Anal. Calcd for C25H16O3: C, 82.40; H, 4.43; Found: C, 82.29; H, 4.71.
2.3.2 Synthesis of 2′,7′-bis(4-nitrophenoxy)-spiro(fluorene-9,9′-xanthene)(2).
In a three-necked flask equipped with a nitrogen inlet and a condenser, 7.28 g (20 mmol) of 1 was first dissolved in 80 ml of DMAc, 5.78 g (42 mmol) of 1-fluoro-4-nitrobenzene and 5.38 g (42 mmol) of potassium carbonate were added subsequently. The mixture was heated at 110 °C for 12 h with stirring under nitrogen, then was poured into 400 mL of solution consisting of equal volumes of ethanol and water, and yellowish solids precipitated out of the solution overnight. After filtration, the residual reactants and potassium carbonate were eliminated from the solids by washing with water, methanol and ethanol consecutively. Finally, 2′,7′-bis(4-nitrophenoxy)-spiro(fluorene-9,9′-xanthene) (2) with solids was collected and dried at 100 °C for 12 h. The crude 2 was purified by recrystallized from acetone to afford white microcrystals (10.54 g, 87%), mp = 163–165 °C by DSC (2 °C min−1).
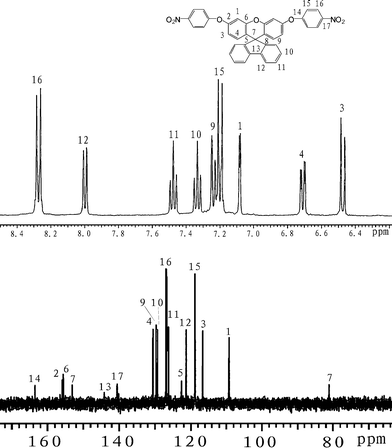
1H NMR (acetone-d6): δ6.46 (d, J = 8.8 Hz, 2H, H3), 6.69 (dd, J = 2.4,2.8 Hz, 2H, H4), 7.08 (d, J = 2.4 Hz, 2H, H1), 7.18 (d, J = 8.4 Hz, 4H, H15), 7.21 (d, J = 7.2 Hz, 2H, H9), 7.31 (dd, J = 7.6,7.2 Hz, 2H, H10), 7.45 (dd, 7.6,7.2 Hz, 2H, H11), 7.99 (d, J = 7.2 Hz, 2H, H12), 8.25 (d, J = 8.4 Hz, 4H, H16). 13C NMR (acetone-d6): 80.4 (C7), 109.2 (C1), 116.6 (C3), 118.7 (C15), 121.2 (C12), 124.4 (C5), 126.2 (C11), 126.8 (C16), 129.2 (C10), 129.5 (C9), 130.4 (C4), 140.5 (C17), 144.1 (C13), 153.0 (C8), 155.5(C6), 155.8 (C2), 163.4 (C14). FT-IR (KBr): 3088–2956 cm−1 (C–H stretch), 1519 and 1348 cm−1(–NO2 stretch), 1260 and 1206 cm−1(C–O stretch). EI-MS (m/z):[M]+ calcd for C37H22N2O7, 606; Found 606. Anal. Calcd for C37H22N2O7: C, 73.26; H, 3.66; N, 4.62; Found: C, 71.29; H, 4.51; N, 4.41.
2.3.3 Synthesis of 2′,7′-bis(4-aminophenoxy)-spiro(fluorene-9,9′ -xanthene)(3).
A mixture of the purified dinitro compound 2 (9.1 g, 15 mmol), 10% Pd/C (0.15 g), ethanol (150 mL), and hydrazine monohydrate (15 mL) was heated at reflux temperature for about 12 h. The resulting clear, darkened solution was filtered hot to remove Pd/C, and the filtrate was then distilled to remove the solvent. The crude product was purified by recrystallization from acetone/water to give pale-white powdery crystals (7.04 g, 86%); mp = 233–235 °C by DSC (2 °C min−1).
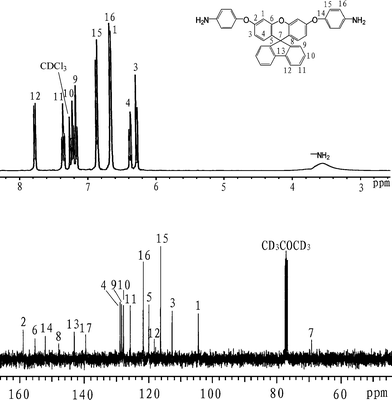
1H NMR (CDCl3): δ3.54 (br, 4H, –NH2), 6.26 (d, J = 8.8 Hz, 2H, H3), 6.46 (dd, J = 6.0,2.4 Hz, 2H, H4), 6.61(d, J = 2.4 Hz, 2H, H1), 6.64 (d, J = 8.4 Hz, 4H, H16), 6.84 (d, J = 8.4 Hz, 4H, H15), 7.14 (d, J = 7.2 Hz, 2H, H9), 7.21(dd, J = 7.6,7.2 Hz, 2H, H10), 7.33 (dd, J = 7.6,7.2 Hz, 2H, H11), 7.75 (d, J = 8.0 Hz, 2H, H12). 13CNMR(CDCl3): δ69.3 (C7), 104.4 (C1), 112.6 (C3), 116.1 (C15), 118.1(C12), 119.8 (C5), 121.5 (C16), 125.6 (C11), 127.7 (C10), 128.3 (C9), 128.7 (C4), 139.5 (C17), 143.6 (C13), 147.8 (C8), 151.9 (C14), 155.1 (C6), 158.8 (C2). FT-IR (KBr): 3428, 3352 cm−1 (N–H stretch), 1248 and 1206 cm−1(C–O stretch). EI-MS (m/z): [M]+ calcd for C37H22N2O3, 546; Found 546. Anal. Calcd for C37H22N2O3: C, 81.30; H, 4.79; N, 5.21; Found: C, 79.69; H, 4.31; N, 5.01.
2.3.4 Synthesis of polyimides.
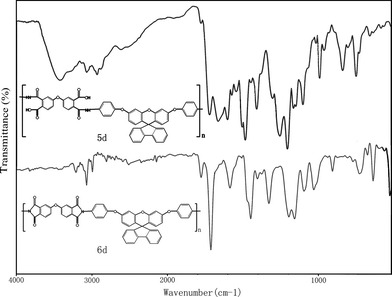
The polyimides were synthesized from various dianhydrides and the diamine3via a conventional two-step method. The synthesis of polyimide 6d is used as an example to illustrate the general synthesis route used to produce the polyimides. To a solution of 0.8199 g (1.50 mmol) of diamine 3 in 15 mL of CaH2–dried DMAc in a 50 mL flask, 0.4653 g (1.50 mmol) of dianhydride ODPA was added in one portion. Thus, the solid content of the solution is approximately 12 wt%. The mixture was stirred at room temperature overnight (for about 24 h) to afford a highly viscous poly(amic acid) solution. The inherent viscosity of the resulting poly(amic acid) 5d was 1.02 dL/g, measured in DMAc at a concentration of 0.5 g/dL at 30 °C. The poly(amic acid) was subsequently converted to polyimide by either thermal or chemical imidization process. For the thermal imidization process, about 4 g of the obtained poly(amic acid) solution was poured into a 5 cm glass culture dish, which was placed overnight in a 90 °C oven to slowly release the casting solvent. The semi-dried poly(amic acid) film was further dried and transformed into polyimide 6d by sequential heating at 150 °C for 30 min, 200 °C for 30 min, and 250 °C for 1 h. The polyimide film was stripped from the glass substrate by being soaked in water. The inherent viscosity of the polyimide 6d was 1.09 dL/g in DMAc at a concentration of 0.5 g/dL at 30 °C. For the tensile test, dielectric and thermogravimetric analyses, the polyimide films were heated at 300 °C for another 1 h. For the chemical imidization process, 3 mL of acetic anhydride and 1 mL of pyridine were added to the remaining poly(amic acid) solution, and the mixture was heated at 100 °C for 1 h to effect a complete imidization. The resulting polyimide solution was poured slowly into 250 mL of methanol giving rise to a fibrous precipitate, which was washed thoroughly with methanol and water, collected by filtration, and dried. The inherent viscosity of chemically imidized6d is 0.81 dL/g in DMAc, measured at a concentration of 0.5 g/dL at 30 °C. FT-IR (film): 1780, 1724 (imide C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch), 1374 (imide C–N stretch), 1261 and 1236 cm−1 (C–O stretch).
O stretch), 1374 (imide C–N stretch), 1261 and 1236 cm−1 (C–O stretch).
3. Results and discussion
3.1 Monomer synthesis
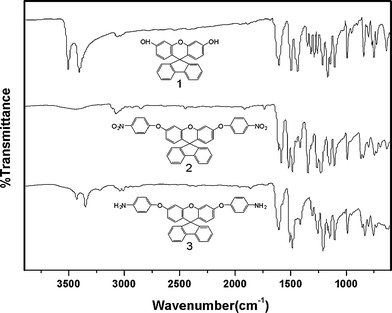
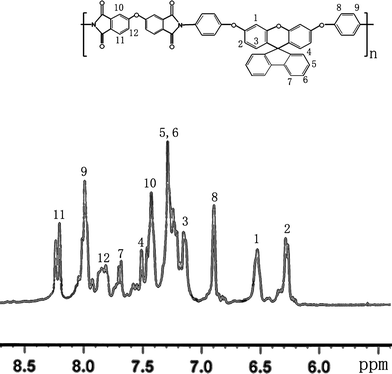
Three steps were used to synthesize the new diamine 3 which contained a spiro(fluorene-9,9′-xanthene) skeleton from 9-fluorenone as shown in Scheme 1. The key intermediate spiro(fluorene-9,9′-(2′,7′-dihydroxyxanthene) (1) was prepared through a simple acid catalyzed condensation reaction9-fluorenone with resorcinol, using ZnCl2/HCl as a condensing agent, to form the spiro framework.44,45 Subsequent the dinitro compound 2 was synthesized by nucleophilic aromatic substitution of 1 with 1-fluoro-4-nitrobenzene in the presence of postassium carbonate as base in DMAc. The diamine3 was obtained in high purity and high yields by the catalyticreduction of intermediate dinitro compound 2 with hydrazine monohydrate and Pd/Ccatalyst in refluxing ethanol. Elemental analysis, IR, and NMR spectroscopic techniques were used to identify structures of the intermediate compounds (1 and 2) and target diamine monomer (3). The FT-IR spectra of compounds 1, 2 and 3 are shown in Fig. 1. Bisphenol compound 1 displayed characteristic absorption bands corresponding to –OH stretching at 3504–3404 cm−1 and –C–O–C– stretching band around 1260 cm−1. Dinitro compound 2 displayed characteristic absorption bands corresponding to asymmetric and symmetric NO2 stretching at 1519 and 1348 cm−1, respectively, which disappeared after reduction. Diamine compound 3 exhibited typical N–H stretching bands at 3428 and 3352 cm−1. Both the dinitro and diamine compounds showed the –C–O–C– stretching band around 1240 cm−1 confirming the presence of the aromatic ether linkage. Fig. 2 display the 1H and 13C NMR spectra of bisphenol compound 1. In the case of Fig. 2, all the aromatic protons of 1 resonated in the region of 6.13–7.88 ppm and the protons of –OH were appeared at farther downfield (8.49 ppm). In 13C NMR spectra, the central spiro carbon (C7) signal resonate at 63.4 ppm indicative of the presence of a spiro skeleton in 1 and the 13 main signals appeared and the number of carbons was consistent with the structure. Fig. 3 and 4 display the 1H and 13C NMR spectra of compounds 2 and 3, respectively. The NMR spectra confirmed that the nitro groups had been converted into amino groups by the high field shift of the aromatic protons and carbons. The signal at 3.54 ppm corresponds to the primary aromatic amineprotons. All the spectroscopic data obtained were in good agreement with the expected structures. The structure of the diamine was designed to impart several desirable properties to polyimides.
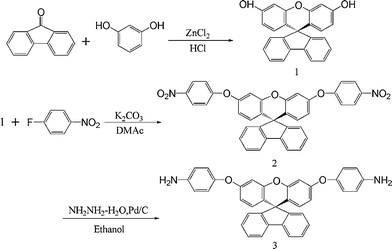 |
| | Scheme 1 Synthesis of the diamine monomer 3. | |
 |
| | Scheme 2 Synthesis of the polyimides. | |
Table 1 Inherent viscosity of poly(amic acid)s and polyimides and thin film tensile properties of the polyimides
|
Poly(amic acid)
|
Polyimide
|
GPC data for polyimides |
Tensile properties of the polyimide films |
| Code |
ηinha (dL/g) |
Code |
ηinha (dL/g) |
M
n
|
M
w
|
Mw/Mn |
Tensile strength (MPa) |
Elongation to break (%) |
Initial modulous (GPa) |
|
Measured at a polymer concentration at 0.5 g/dL in DMAc at 30 °C.
Insoluble in DMAc.
Insoluble in THF.
|
|
5a
|
0.87 |
6a
|
—b |
—c |
|
|
76 |
7 |
1.78 |
|
5b
|
0.79 |
6b
|
0.92 |
—c |
|
|
80 |
7 |
1.76 |
|
5c
|
0.97 |
6c
|
1.13 |
23,000 |
45,000 |
1.95 |
112 |
10 |
2.06 |
|
5d
|
1.02 |
6d
|
1.09 |
24,000 |
47,500 |
2.07 |
124 |
11 |
2.11 |
|
5e
|
0.92 |
6e
|
0.69 |
21,000 |
41,500 |
1.97 |
92 |
9 |
2.10 |
|
Polyimide
|
Formula of the repeat unit (formula weight) |
C
(%) |
H
(%) |
N
(%) |
| Calcd. |
Found |
Calcd. |
Found |
Calcd. |
Found |
|
6a
|
C47H24N2O7(728.71) |
77.25 |
76.78 |
3.42 |
3.21 |
3.91 |
3.78 |
|
6b
|
C53H28N2O7(804.81) |
78.91 |
78.56 |
3.73 |
3.54 |
3.45 |
3.55 |
|
6c
|
C54H28N2O8(832.82) |
77.72 |
77.41 |
3.58 |
3.46 |
3.37 |
3.21 |
|
6d
|
C53H28N2O8(820.74) |
77.36 |
77.17 |
3.64 |
3.38 |
3.41 |
3.53 |
|
6e
|
C56H28F6N2O7(954.84) |
70.31 |
70.13 |
3.08 |
3.24 |
2.97 |
3.68 |
3.3 Properties of polyimides
3.3.1 Organo-solubility and film property.
The solubility of the polyimides was tested qualitatively in various organic solvents. The solubility properties of the thermally cured polyimides are reported in Table 3. All the polyimides were soluble in dipolar amide-type solvents, such as NMP, DMAc, and DMF, at room temperature or upon heating. Some of them also were soluble in DMSO and phenolsolvents like m-cresol and o-chlorophenol, as well as chlorinated solvents like chloroform. The polyimides (6d–6e) derived from less stiff dianhydride components were also soluble in low boiling point organic solvents such as THF and 1,4-dioxane. In general, the present spiroskeletons linkage 6 series polyimides revealed an enhanced solubility, and this could be attributed to the presence of kinked spirofluorene units, with flexible aryl ether linkages along the polymer backbone, which increased the disorder in the chains and hindered dense chain packing, thus, reducing the interchain interactions to enhance the solubility. The solubilities of the resulting polyimides 6a–6e by chemical imidization were also investigated, and the result is presented in Table 3. The polyimide prepared by chemical imidization exhibited higher solubility than those by thermal curing. The less solubility of the latter is possibly due to the presence of partial interchain crosslinking or denser aggregation of the polymer chains during the thermal imidization process. All of the spiroskeletons linkage polyimides afforded good quality and tough films with light color. These films were subjected to a tensile test, and their tensile properties are also summarized in Table 1. The films exhibited ultimate tensile strengths of 76–124 MPa, elongations to break of 7–11%, and initial moduli of 1.76–2.11 GPa, indicating that they are strong and tough polymeric materials. The crystallinity of the polyimides was characterized by wide-angle X-ray diffraction (WAXD) studies. Two of the PI 6 series, 6a and 6b, displayed slightly semi-crystalline WAXD patterns, whereas all of the others showed amorphous patterns. The amorphous nature of the polyimides 6a–6e is attributed to the spiroskeletons structure and flexible ether linkage coming from the diamine monomer.
Table 3 Solubility behavior of the polyimides prepared via thermal imidization and chemical imidization a,b
|
Solvent
c
|
Polyimides |
|
6a
|
6b
|
6c
|
6d
|
6e
|
|
The symbol +: soluble at room temperature; +h: soluble on heating at 100 °C or boiling temperature; −: insoluble even on heating.
The solubility was determined by using 10 mg sample in 1 mL of stirred solvent.
NMP = N-methyl-2-pyrrolidone; DMAc = N-dimethylacetamide; DMF = N-dimethylformamide; DMSO = dimethyl sulfoxide; THF = tetrahydrofuran.
Data in parentheses are those for chemical imidization polyimides.
|
|
NMP
|
+h(+)d |
+h(+) |
+ (+) |
+(+) |
+(+) |
|
DMAc
|
+h(+h) |
+h(+h) |
+h(+) |
+(+) |
+(+) |
|
DMF
|
+h(+h) |
−(+h)
|
+h(+) |
+(+) |
+(+) |
|
DMSO
|
−(−) |
−(−) |
+h(+h) |
+ (+) |
+(+) |
|
m-Cresol
|
−(+h)
|
+h(+) |
+h(+) |
+(+) |
+(+) |
|
o-Chorophenol
|
−(−) |
+h(+) |
+(+) |
+(+) |
+(+) |
|
THF
|
−(−) |
−(−) |
+(+) |
+(+) |
+(+) |
|
Pyridine
|
−(−) |
−(−) |
−(+) |
+h(+) |
+h(+) |
|
1,4-Dioxane
|
−(−) |
−(−) |
−(+) |
+(+) |
+(+) |
|
chloroform
|
−(−) |
−(−) |
+h(+) |
+h(+) |
+(+) |
| CH2Cl2 |
−(−) |
−(−) |
−(h+)
|
+h (+h) |
+h (+) |
|
Acetone
|
−(−) |
−(−) |
−(−) |
−(+h)
|
−(+h)
|
3.3.2 Thermal properties.
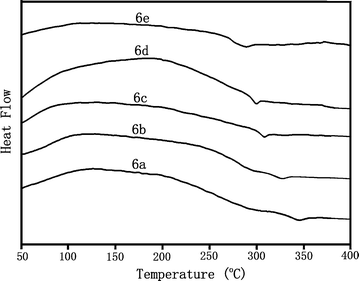
The thermal properties of the polyimides were determined by using DSC and TGA (Table 4). DSC experiments were conducted at a heating rate of 20 °C min−1 in nitrogen. Rapid cooling from 400 °C to room temperature produced predominantly amorphous samples, so the Tg of almost all the polyimides could be easily taken from the second DSC heating traces. DSC curves for the polyimides are reproduced in Fig. 7. The Tg values of the PI 6 series ranged from 296 °C to 345 °C, which increased in the order of PMDA > BPTA > BTDA > 6FDA > OPDA. 6d showed the lowest Tg because of the presence of two flexible ether linkages between the phthalimide units. As in Table 4, the Tgs of PI 6 series are high, demonstrating the Tg-enhancement effect of spiro framework, which will hide the rotation of polymer chains. This indicates that introducing the bulky fluorenegroups, does not sacrifice the glass transition even though the free volume of PI 6 series is larger. The thermal stability of the polyimides was evaluated by TGA measurements in both air and nitrogen atmospheres. Typical TGA curves for polyimide 6d is reproduced in Fig. 8. The decomposition temperatures (Td) at 5% and 10% weight loss in nitrogen and in air atmospheres were determined from the original TGA thermograms and are also given in Table 4. The Td at 10% weight loss of the polyimides (6a–6e) in nitrogen and air stayed in the range of 542–582 °C and 536–570 °C, respectively. They left more than 54% char yield at 800 °C in nitrogen. The TGA data indicated that these polyimides had fairly high thermal stability with the introduction of the spiro framework structure, even though they revealed high solubility and optical transparency.
Table 4 Thermal behavior data and cutoff wavelength (λ0) from UV-Vis spectra of polyimides
|
Polymer
|
T
g
/°C |
T
5
/°C |
T
10
/°C |
Char Yieldc (%) |
λ
0/nm |
| In N2 |
In air |
In N2 |
In air |
|
Glass transition temperature (Tg) was measured by DSC at heating rate of 20 °C min−1 in nitrogen.
Decomposition temperatures recorded by TGA at a heating rate of 20 °C min−1.
Residual weight percentage at 800 °C in nitrogen.
|
|
6a
|
346 |
548 |
540 |
557 |
555 |
54 |
409 |
|
6b
|
331 |
565 |
560 |
582 |
570 |
58 |
378 |
|
6c
|
311 |
541 |
535 |
563 |
551 |
58 |
362 |
|
6d
|
296 |
536 |
524 |
542 |
540 |
54 |
356 |
|
6e
|
304 |
527 |
514 |
544 |
536 |
56 |
352 |
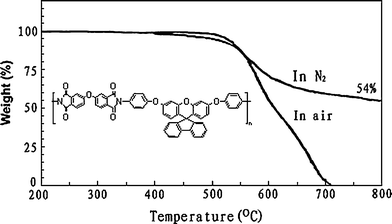 |
| | Fig. 8
TGA curves of polyimide 6d at a heating rate of 20 °C min−1. | |
3.3.3 Color and optical transparency.
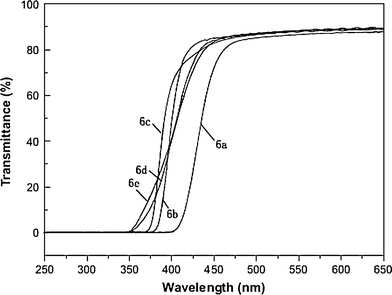
Thin films were measured for optical transparency with UV-visible sepectroscopy. Fig. 9 shows the UV-visibleabsorption spectra of the PI film prepared via thermal imidizations, and cutoff wavelength (absorption edge, λ0) value are listed in Table 4. In agreement with the results obtained from the colorimeter, all the polyimides revealed low λ0 and high optical transparency, with a percentage of transmittance higher than 80% at 450 nm. Because of the highly conjugated aromatic structures and intermolecular charge-transfer complex (CTC) formation of PI, most polymers between the UV and visible area have strong absorption. However, these PIs which have bulky spiroskeletons structure and flexible ether linkage in the center of diamine reduced the intermolecular CTC between alternating electrondonor (diamine) and electron-acceptor (dianhydride) moieties. The polyimides 6d and 6e produced from ODPA and 6FDA were essentially colorless and showed relatively lower λ0 values in contrast to other dianhydrides, and these can be explained by the decreased intermolecular interactions.46 The decrease in chain–chain CTC formation also was understandable from the significant solubility of the polyimides prepared from the spiroskeletons structure diamine3.
3.3.4 Dielectric constant and water absorption.
The dielectric constants and water absorption of the 6 series were measured, and the results were summarized in Table 5. The 6 series showed dielectric constants in the range 2.65–3.12 at 1 KHz, which were much lower than the standard PI such as Kapton films (3.5 at 1 kHz) and Ultem (3.5 at 1 kHz).476c had a higher dielectric constant for the reason that the carbonyl bridge (CO) had less free volume, more polarizability in the PI backbone when compared to the –O– bridge (6d). The low dielectric constant of the 6 series is mainly attributed to the fact that the introduction of the spiro framework and noncoplanar structure loosened the polymer packing, subsequently reducing their density and dielectric constants. As expected, the 6 series also exhibited low water absorptions (0.36–0.73%) due to the water proofing fluorenegroups. Because of the proofing effect of the trifluoromethyl groups, 6e had significantly lower moisture absorption than the other 6 series.
Table 5 Moisture-absorption and dielectric constants of polyimides
|
Polymer
|
Film thickness/μm |
Moisture absorption(%)a |
Dielectric constant |
| 1 KHz |
|
Moisture absorption of polyimide films was measured immersing the films in distilled water at 25 °C for 100 h.
|
|
6a
|
45 |
0.61 |
3.10 |
|
6b
|
53 |
0.68 |
3.04 |
|
6c
|
32 |
0.73 |
3.12 |
|
6d
|
51 |
0.54 |
2.92 |
|
6e
|
36 |
0.36 |
2.65 |
4. Conclusions
A novel spiro(fluorene-9,9′-xanthene) skeleton bis(ether amine) monomer, 2′,7′-bis(4-aminophenoxy)-spiro(fluorene-9,9′-xanthene), was successfully synthesized and characterized in the present work, which was employed in polycondensation with various aromatic dianhydrides, to prepare a series of organosoluble aromatic polyimides. The resulting polyimides could be cast into flexible and strong films with high solubility, high optical transparency, excellent thermal stability, moderate to high glass transition temperatures (296–346 °C), and low dielectric constants. Thus, this series of polyimides exhibits a good combination of properties required for high-performance materials and demonstrates a promising potential for future applications.
Acknowledgements
The authors are grateful for the research support from the Natural Science Foundation of Gansu Province (No. 096RJZA047) and the State Key Laboratory of Applied Organic Chemistry of People's Republic of China.
Notes and references
-
Polyimides, ed. D. Wilson, H. D. Stenzenberger and P. M. Hergenrother, Blackie, Glasgow and London, 1990 Search PubMed.
- C. E. Sroog, Prog. Polym. Sci., 1991, 15, 551–594.
-
Polyimides: Fundamentals and Applications, ed. M. M. Ghosh, K. L. Mittal, Marcel Dekker, New York, 1995 Search PubMed.
- C. W. Chang, H. J. Yen, K. Y. Huang, J. M. Yeh and G. S. Liou, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7937–7949 CrossRef CAS.
- A. S. Mathews, D. J. Kim, Y. K. Kim, I. Kim and C. S. Ha, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 8117–8130 CrossRef CAS.
- G. Y. Lee, H. N. Jang and J. Y. Lee, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3078–3087 CrossRef CAS.
- C. P. Chang, Y. Y Su and Y. C. Chen, Eur. Polym. J., 2006, 42, 721–732 CrossRef.
- J. G. Liu, Y. Nakamura, Y. Shibasaki, S. Ando and M. Ueda, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5506–5517.
- G.-J. Shin, C.-J. Jung, J.-H. Chi, T.-H. Oh and J.-B. Kim, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 776–788 CrossRef CAS.
- M. R. Bellomo, G. Di Pasquale, A. La Rosa, A. Pollicino and G. Siracusa, Polymer, 1996, 37, 2877–2888 CrossRef CAS.
- S. H. Hsiao and P. C. Huang, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 2421–2429 CrossRef CAS.
- D. J. Liaw, B. Y. Liaw and K. L. Su, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 1997–2003 CrossRef CAS.
- J. F. Espeso, E. Ferrero, J. G. De La Campa, A. E. Lozano and J. De Abajo, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 475–485 CrossRef CAS.
- S. H. Hsiao and C. F. Chang, J. Polym. Sci., Part A: Polym. Chem., 1996, 34, 1433–1441 CrossRef CAS.
- C. L. Chung and S. H. Hsiao, Polymer, 2008, 49, 2476–2485 CrossRef CAS.
- C. P. Yang, Y. Y. Su and S. H. Hsiao, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 3140–3152 CrossRef CAS.
- G. C. Eastmond, M. Gibas and J. Paprotny, Eur. Polym. J., 1999, 35, 2097–2105 CrossRef CAS.
- Y. T. Chern and H. C. Shiue, Macromolecules, 1997, 30, 4646–4651 CrossRef CAS.
- L. J. Mathias, A. V. G. Muir and V. R. Reichert, Macromolecules, 1991, 24, 5232–5233 CrossRef CAS.
- C. Y. Wang, G. Li, X. Y. Zhao and J. M. Jiang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3309–3317 CrossRef CAS.
- T. Matsuura, Y. Hasuda, S. Nishi and N. Yamada, Macromolecules, 1991, 24, 5001–5005 CrossRef CAS.
- F. Li, S. Fang, J. J. Ge, P. S. Honigfort, J. C. Chen, F. W. Harris and S. Z. D. Cheng, Polymer, 1999, 40, 4571–4583 CrossRef CAS.
- F. Li, S. Fang, J. J. Ge, P. S. Honigfort, J. C. Chen, F. W. Harris and S. Z. D. Cheng, Polymer, 1999, 40, 4987–5002 CrossRef CAS.
- S. H. Hsiao, C. P. Yang and C. Y. Yang, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 1487–1497 CrossRef CAS.
- D. S. Reddy, C. F. Shu and F. I. Wu, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 262–268 CrossRef CAS.
- J. Salbeck, N. Yu, J. Bauer, F. Weissöortel and H. Bestgen, Synth. Met., 1997, 91, 209–213 CrossRef CAS.
- N. Johansson, J. Salbeck, J. Bauer, F. Weissoörtel, P. Broöms, A. Andersson and W. R. Salaneck, Adv. Mater., 1998, 10, 1136–1141 CrossRef CAS.
- F. Steuber, J. Staudigel, M. Stoössel, J. Simmerer, A. Winnacker, H. Spreitzer and J. Wei, Adv. Mater., 2000, 12, 130–133 CrossRef CAS.
- J. H. Weisburger, E. K. Weisburger and F. E. Ray, J. Am. Chem. Soc., 1950, 72, 4250–4253 CrossRef CAS.
- R. Wu, J. S. Schumm, D. L. Pearson and J. M. Tour, J. Org. Chem., 1996, 61, 6906 CrossRef CAS.
- D. Katsis, Y. H. Geng, J. J. Ou, S. W. Culligan, A. Trajkovska, S. H. Chen and L. J. Rothberg, Chem. Mater., 2002, 14, 1332–1339 CrossRef CAS.
- K. T. Wong, Y. Y. Chien, R. T. Chen, C. F. Wang, Y. T. Lin, H. H. Chiang, P. Y. Hsieh, C. C. Wu, C. H. Chou, Y. O. Su, G. H. Lee and S. M. Peng, J. Am. Chem. Soc., 2002, 124, 11576–11577 CrossRef CAS.
- W. L. Yu, J. Pei, W. Huang and A. J. Heeger, Adv. Mater., 2000, 12, 828–834 CrossRef CAS.
- D. Marsitzky, J. Murray, J. C. Scott and K. R. Carter, Chem. Mater., 2001, 13, 4285–4292 CrossRef CAS.
- F. I. Wu, R. Dodda, D. S. Reddy and C. F. Shu, J. Mater. Chem., 2002, 12, 2893–2901 RSC.
- C. L. Chiang and C. F. Shu, Chem. Mater., 2002, 14, 682–687 CrossRef CAS.
- S. H. Hsiao and C. T. Li, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 1403–1412 CrossRef CAS.
- C. H. Chou, D. S. Reddy and C. F. Shu, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3615–3621 CrossRef CAS.
- C. P. Yang, Y. Y. Su, S. J. Wen and S. H. Hsiao, Polymer, 2006, 47, 7021–7033 CrossRef CAS.
- C. Y. Wang, X. Y. Zhao, G. Li and J. M. Jiang, Polym. Degrad. Stab., 2009, 94, 1746–1753 CrossRef CAS.
-
S. Yoshida, T. Fujimori and N. Kato, Jpn. Pat., 57915, 2007.
- R. Mercado, Y. Wang, T. Flaim, W. DiMenna and U. Senapati, Proc. SPIE, 2004, 5351, 276 CAS.
- C. A. Terraza, J. G. Liu, Y. Nakamura, Y. Shibasaki, S. Ando and M. Ueda, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1510–1520 CrossRef CAS.
- F. Bischoff and H. J. Adkins, J. Am. Chem. Soc., 1923, 45, 1030–1033 CrossRef CAS.
- L. H. Xie, F. Liu, C. Tang, X. Y. Hou, Y. R. Hua, Q. L. Fan and W. Huang, Org. Lett., 2006, 8, 2787–2790 CrossRef CAS.
- S. Ando, T. Matsuura and S. Sasaki, Polym. J., 1997, 29, 69–75 CrossRef CAS.
- S. Banerjee, M. K. Madhra and V. Kute, J. Appl. Polym. Sci., 2004, 93, 821–828 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.