Ambidextrous circular dichroism and circularly polarised luminescence from poly(9,9-di-n-decylfluorene) by terpene chirality transfer†
Yoko
Nakano
a,
Yang
Liu
b and
Michiya
Fujiki
*a
aGraduate School of Materials Science, Nara Institute of Science and Technology, 8916-5 Takayama, Ikoma, Nara 630-0192, Japan. E-mail: fujikim@ms.naist.jp; Tel: +81-743-72-6040
bSuzhou Institute of Nano-tech and Nano-bionics (SINANO), Chinese Academy of Sciences, Dushu Lake Higher Education Town, Ruoshui Road 398, Suzhou Industrial Park, Suzhou 215125, China. E-mail: yliu2007@sinano.ac.cn; Tel: +86-512-6287-2546
First published on 22nd December 2009
Abstract
Solvent chirality transfer using enantiomeric pairs of limonene and α-pinene allowed for the successful production of optically active poly(9,9-di-n-decylfluorene) (PF10) aggregates with circular dichroism (CD) and circularly polarised luminescence (CPL) properties. The aggregates were rapidly produced by CD-/CPL-silentPF10 with the aid of solvent chirality transfer at 25 °C. We demonstrate that: (i) through weak forces, such as π/π, van der Waals, and CH/π interactions, CD-/CPL-active PF10 aggregates successfully emerge in a chiral tersolvent system of chloroform (as a good solvent), alkanol (as a poor solvent), and terpenes (as a chiral solvent), (ii) the molecular weight of PF10, alkyl chain length of PFs, terpene structure and enantiopurity, solvent type, and solution temperature greatly affect the magnitude and sign of the CD-/CPL-signals, (iii) the vortex and aggregate size only mildly affect the magnitude of these signals, and (iv) a new CPL band at 434 nm from PF10 aggregates, arising from the CD/UV-vis band at 428 nm, may result from a chiral β-phase. The terpene chirality transfer demonstrated herein may provide a new environmentally friendly, safer, and milder process to rapidly produce ambidextrous chiroptical polymeric materials at ambient temperature from CD-/CPL-silentpolymers without any specific chiral catalysts or substituents.
Introduction
Since the time of Pasteur, the origin of biomolecular handedness on earth has remained a mystery to scientists.1 Nature's elegant bottom–up preference has led to the development of synthetic polymers,2 supramolecules,3 liquid crystals,4 small molecules,5 and organic solid crystals,6 which have (chir)optical signals that enable us to study them through their ambidextrous circular dichroism (CD) and circularly polarised luminescence (CPL) properties. Such materials can be produced under mild and ambient conditions. Among these artificial substances, chromophoric and fluorophoric optically active polymers in the form of film states and aggregates have recently received a great deal of attention for their applications in optical data processing and display devices.2b,3a They show intense CD signals associated with an efficient CPL emission in the UV-vis region, properties that may be promising for circular polarisation-based optical devices.Among a number of the π- and σ-conjugating polymers, polyfluorene (PF) derivatives showing excellent thermal, chemical, and photochemical stabilities have several advantages for the study of their (chir)optical properties due to their intense blue-colour photoluminescence (PL) and/or electroluminescence with a high quantum yield.7,8 After obtaining an optically active PF, the most common initial approach is to introduce chiral substituents to the 9,9-position of the fluorene rings.8b–8e,8g,8h These systems allow helical organisations to yield amplified supramolecular chirality through different types of noncovalent forces, including π–π stacking, hydrogen bonding, and solvophobic and ionic interactions. These classical forces permit the molecular chirality to be transferred to the chiral supramolecular structures,8 and these artificial helical architectures can endow various functional materials with unique chiroptical properties.
Recent knowledge of nonclassical forces has led to a better understanding of various weak/ultraweak forces,9–11 including van der Waals (∼1 kcal mol−1), π/π (∼1 kcal mol−1), CH/π (∼0.5 kcal mol−1), CH/O (∼0.5 kcal mol−1), and CF/Si forces (∼0.001 kcal mol−1),9d which play key roles in constructing the higher-order structures of biopolymers, artificial polymers, supramolecules, and molecular crystals. These weak and ultraweak forces offer unlimited opportunities for designing and synthesising optically active π-conjugated polymers that exhibit enhanced CPL and CD properties from optically inactive (CD-silent) polymers through the transfer of molecular chirality. In the case of intense acid–base interactions (∼10 kcal mol−1), the formation of a one-handed helix is possible from CD-silent polyphenylacetylene carrying a carboxyl moiety (∼10−2 mol as the repeating unit per litre) and can be induced by the addition of an almost equal amount of chiral molecular amine (∼10−2 mol per litre) through intermolecular chiral proximity forces.10 However, if the intermolecular chiral forces are insufficient in solution, a higher concentration of chiral molecules or different solvent is needed.2d
Previous studies on the chiral solvent effect have shown that a proper choice of a poorer, polar chiral solvent and/or an appropriate volume fraction in a polar chiral-achiral cosolvent is required to effectively induce a helical conformation from CD-silent systems, including optically inactive, achiral, and racemic substances.12 The emergence of Cotton CD signals due to the twisted forms of CD-silent benzyl and benzophenone dissolved molecularly in (2S,3S)–butanediol was the first example of the chiral solvent effect.12a A cooperative helical preference revealed by the CD characteristics of poly(n-hexylisocyanate), which consists of polar amides in the backbone dissolved in polar chlorinated chiral solvents, may be the second example of the chiral solvent effect and the first example for chain-like polymers.12b A further study for polyisocyanate showed that helix formation was effectively induced when a poor “precipitating” solvent was employed as a cosolvent in combination with a second, chiral solvent.12c
In a previous paper, we reported on the addition of methanol (an achiral, poor, polar solvent) to a CD-silent polysilane carrying an achiral ether moiety as the substituent, which was dissolved in a mixture of chiral alcohol and tetrahydrofuran (achiral, good, polar cosolvent). An optically active polysilane aggregate was successfully produced through a relatively weak OH/O interaction (∼2 kcal mol−1) between the polymer and solvent molecules.2d An optically active polymer aggregate can also be produced through solvent chirality transfer if a poorer polar solvent system is appropriately chosen. However, these synthetic chiral solvents are usually expensive and not common for organic synthesis. In addition, polar functional groups for both the solvent molecule and polymer structure may be required to effectively mediate the interaction between the solvent chirality and CD-silentpolymer if only XH/O-type forces are applied.
If an enantiomeric pair of nonpolar chiral solvent molecules and nonpolar CD-silentpolymers effectively interact with each other via weak forces including π/π, van der Waals, and CH/π interactions, the production of a broad range of ambidextrous chiroptical polymers in the form of films and/or aggregates is possible. To test this method, we used limonenes and pinenes as nonpolar terpenes, inexpensive naturally occurring biomass enantiomers as the chiral solvent and poly(9,9-di-n-alkylfluorene)s as a model nonpolar CD-/CPL-silent chromophoric and fluorophoric polymer.
In this article, we report (Chart 1) that: (i) through weak forces, including π/π, weak van der Waals, and CH/π interactions, CD-/CPL-active poly(9,9-di-n-decylfluorene) (PF10) aggregates were successfully produced in a chiral tersolvent system of chloroform (as a good solvent), methanol (as a poor solvent), and limonene (as a chiral solvent); (ii) the molecular weight of PF10, n-alkyl chain length of the PFs, terpene structures, enantiopurity of limonene, solvent type, and solution temperature greatly affected the magnitude of the optical activity of the PF10; (iii) the vortex and aggregate size of the optically active PF10 only weakly affected the magnitude of its optical activity; and (iv) a new CPL band at 434 nm from the PF10 aggregates, arising from CD signals at 428 nm, may have resulted from a chiral β-phase.
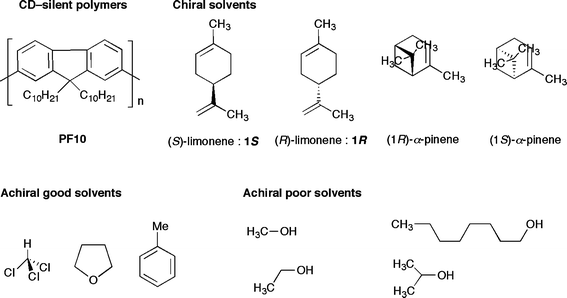 | ||
| Chart 1 Chemical structures of poly(9,9-di-n-decylfluorene) (PF10), limonenes (1R and 1S), α-pinenes, and achiral solvents. | ||
Experimental section
Instrumentation
The CD/UV-vis spectra of the solution were recorded simultaneously at 25 °C on a JASCO J-820 spectropolarimeter equipped with a Peltier-controlled housing to control the stir speed, direction, and temperature. An SQ-grade quartz cuvette was used with a path length of 10 mm, a scanning rate of 100 nm min−1, a bandwidth of 1 nm, a response time of 1 s, and single accumulation. The CPL/PL spectra were recorded at 25 °C on a JASCO CPL-200 spectrofluoropolarimeter equipped with a Peltier-controlled housing and an SQ-grade quartz cuvette. A path length of 10 mm, a scanning rate of 100 nm min−1, a bandwidth for excitation of 10 nm, a bandwidth for monitoring of 10 nm, a response time of 2 s, and single accumulation were employed. This instrument was manipulated to obtain a high S/N ratio by adjusting the angle between the incident and travelling light to 0° using a notch filter. The PL quantum yield was determined relative to 9,10-diphenylanthracene (Φ = 90%, in cyclohexane).13 The optical rotation at the Na-d line was measured with a JASCO P-1020 polarimeter using a path length of 10 mm at room temperature. Chiral gas chromatography (Shimadzu GC-2010), using a Supelco β-DEX-120 with 30 m × 0.25 mm ID, was used to determine the enantiopurity of the limonenes. A column oven temperature program was used to set an initial temperature of 40 °C and a heating rate 20 °C per min and then hold at 85 °C for 20 min, and He at a flow rate of 1.2 mL min−1 served as the carrier gas. The 1H and 13C NMR spectra were recorded with a JEOL EX-400 spectrometer at 400 MHz CDCl3 and 24 °C. The weight-average molecular weight (Mw), number-average molecular weight (Mn), and polydispersity index (PDI = Mw/Mn) were evaluated by gel permeation chromatography (GPC) at 40 °C on a Shimadzu A10 instrument with a PLgel (Varian) 10 μm mixed-B column. HPLC-grade chloroform was used as the eluent, and the measurements were calibrated using polystyrene standards (Varian). A fluorescent optical micrograph (FOM) excited at 365 nm was taken with a Nikon Eclipse E400 optical microscope equipped with a Nikon CCD camera. Wide-angle X-ray diffraction (WAXD) measurements were performed with a Rigaku Ultra X18 instrument using an X-ray wavelength of 1.54 Å, Cu Kα radiation, and a Ni filter.Chiroptical analysis14
The magnitude of the circular polarisation at the ground state is defined as gCD = 2 × (εL − εR)/(εL + εR), where εL and εR denote the extinction coefficients for left and right circularly polarised light, respectively. The magnitude of circular polarisation of the excited state is defined as gCPL = 2 × (IL − IR)/(IL + IR), where IL and IR indicate the output signals for left and right circularly polarised light. Experimentally, the value of gCD is defined as Δε/ε = [ellipticity/32,980]/absorbance at the CD extremum, and the value of gCPL is defined as ΔI/I = [ellipticity/(32,980/ln10)]/unpolarised PL intensity at the CPL extremum.Materials
Spectroscopic grade chloroform and methyl alcohol (Dotite) were used to prepare the polymer solutions for measurements. (R)-limonene (1R) and (S)-limonene (1S) were obtained from Wako (Tokyo, Japan) and purified by reduced pressure prior to use. 1R: [α]27589 = +101.55° (neat), >99.1% ee; 1S: [α]27589 = −103.19° (neat), >99.3% ee. (1R)- and (1S)-α-pinenes were purchased from Tokyo Chemical (Tokyo, Japan) and used as received. A series of poly(9,9-di-n-alkylfluorene)s (PFs) was produced according to a synthetic approach previously described (ESI, Scheme S1†).8b–8i The corresponding monomers, 2,7-dibromo-9,9-di-n-alkylfluorenes, were prepared by double alkylation of 2,7-dibromofluorene (Aldrich) at the C-9 position in the presence of a strong base and a phase transfer catalyst. The pure products were obtained by purification using silicacolumn chromatography after recrystallisation from ethanol. The PFs with various alkyl side groups were synthesised by Yamamoto's coupling polymerisation of 2,7-dibromo-9,9-di-n-alkylfluorene in the presence of Ni(COD)2 (Acros and Kanto), which can potentially generate polymers with weight-average molecular weights (Mw) up to 300,000. The polymeric products were obtained by filtration through a PTFEmembrane filter (0.45 μm pore) and precipitation in methanol (twice). The structures of the monomers and polymers were characterised by 1H and 13C NMR spectroscopy.8g–8i The CD and UV spectra of 1S and 1R in n-hexane at 25 °C (Fig. S1†) indicated that their signals do not interfere with the chiroptical CD and UV spectra of PFs and their aggregates between 225 and 800 nm, even in the chiral tersolvent system.Preparation of optically active PF aggregates
To survey whether an optically active PF can be produced with this method, we first used a combinatorial approach with a mixture of chiral, poor, and good solvents to find the optimised volume fractions. Among the polymers produced by solvent chirality transfer, the optically active PF10-150 K (Mw = 1.48 × 105, PDI = 3.58) aggregates were of particular interest. The total volume of the mixed solvent, or the mixture of methanol and chloroform, was fixed at 3.0 mL. The amount of CD-active PF10-150 K aggregates varied with the relative volume fraction of 1S and methanol (chloroform: fixed) (Fig. S2a†), the relative volume fraction of 1S (chloroform + methanol: fixed) (Fig. S2b†), and the relative volume fraction of 1S and chloroform (methanol: fixed) (Fig. S2c†). This set-up was similar to the other chiral tersolvent systems. The marked changes in the UV-vis spectra of CD-inactive PF10-150 K aggregates were observed when varied with the relative volume fraction of methanol and chloroform in the absence of limonene (Fig. S2d†).A typical procedure for the production of PF aggregates in a mixed chloroform/limonene/methanolsolvent is described below. First, 0.7 mL of 1S was added to 0.3 mL of a chloroform stock solution containing PF (2.5 × 10−4 mol L−1 of the fluorene repeating unit for the CD/UV-vis studies, 5.0 × 10−5 mol L−1 of the fluorene repeating unit for the CPL/PL studies) in the SQ-cuvette, which was placed in the Peltier apparatus of a JASCO J-820 spectropolarimeter and CPL-200 spectrofluoropolarimeter at 25 °C and stirred clockwise for 10 s at 800 rpm. We then added 2.0 mL of methanol at 25 °C, resulting in a white turbid solution of PF aggregates dispersed in the solvent. After stirring for 30 s, this solution was used for CD/UV-vis and CPL/PL spectroscopic measurements.
Results and discussion
Emergence of cotton CD signals from CD-silentPF10
Typical CD and UV-vis spectra of ambidextrous PF10-150 K aggregates formed in chiral tersolvents at 25 °C under the optimised volume fraction composed of limonene/chloroform/methanol: 0.7/0.3/2.0 (v/v/v) are shown in Fig. 1a. No detectable CD signals in the region of the π–π* transition of PF10-150 K were recorded in either the molecularly dispersed form in chloroform (a good achiral solvent) or in limonene/ chloroform (a good chiral cosolvent) (Fig. S3a–S3b†) since no efficient chiral π/π, CH/π, or van der Waals interactions were present between the n-decyl substituent of PF10-150 K and the two isolated double bonds of limonene.![(a) Typical CD and UV-vis spectra (a (): 1S, b (⋯): 1R) of ambidextrous CD-active PF10-150 K aggregates formed in a chiral tersolvent at 25 °C under the optimised volume fraction of limonene/chloroform/methanol: 0.7/0.3/2.0 (v/v/v), [FL repeating unit] = 2.5 × 10−5 mol L−1, with stirring at 800 rpm (CW). (b) CD and UV-vis spectra of CD-silent PF10-150 K aggregates formed in an achiral cosolvent at 25 °C under the optimised volume fraction of chloroform/methanol: 1.5/1.5 (v/v), [FL repeating unit] = 2.5 × 10−5 mol L−1, and 800 rpm stir rate (CW).](/image/article/2010/PY/b9py00288j/b9py00288j-f1.gif) | ||
Fig. 1 (a) Typical CD and UV-vis spectra (a (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ): 1S, b (⋯): 1R) of ambidextrous CD-active PF10-150 K aggregates formed in a chiral tersolvent at 25 °C under the optimised volume fraction of limonene/chloroform/methanol: 0.7/0.3/2.0 (v/v/v), [FL repeating unit] = 2.5 × 10−5 mol L−1, with stirring at 800 rpm (CW). (b) CD and UV-vis spectra of CD-silent PF10-150 K aggregates formed in an achiral cosolvent at 25 °C under the optimised volume fraction of chloroform/methanol: 1.5/1.5 (v/v), [FL repeating unit] = 2.5 × 10−5 mol L−1, and 800 rpm stir rate (CW). ): 1S, b (⋯): 1R) of ambidextrous CD-active PF10-150 K aggregates formed in a chiral tersolvent at 25 °C under the optimised volume fraction of limonene/chloroform/methanol: 0.7/0.3/2.0 (v/v/v), [FL repeating unit] = 2.5 × 10−5 mol L−1, with stirring at 800 rpm (CW). (b) CD and UV-vis spectra of CD-silent PF10-150 K aggregates formed in an achiral cosolvent at 25 °C under the optimised volume fraction of chloroform/methanol: 1.5/1.5 (v/v), [FL repeating unit] = 2.5 × 10−5 mol L−1, and 800 rpm stir rate (CW). | ||
However, optically active PF10-150 K aggregates formed from CD-silentPF10-150 K in the chiral tersolvent condition and exhibited an intense CD spectrum with three distinct CD extrema in the region of the π–π* transitions. When the 1R tersolvent system was used in place of the 1S tersolvent system, an almost mirror image inversion of the Cotton CD spectrum for the PF10-150 K aggregate occurred, as expected. The PF10-150 K aggregates had extremely high quantum yields at 25 °C, regardless of the limonene chirality. The values were corrected by an average refractive index of the mixed solvent: Φ = 53% (excited at 372.6 nm) in 1S/chloroform/methanol (0.7/0.3/2.0 (v/v/v)), Φ = 54% (excited at 372.6 nm) in 1R/chloroform/methanol (0.7/0.3/2.0 (v/v/v)), and Φ = 59% in chloroform–methanol (1.5/1.5 (v/v)).
The magnitude and extrema of the three major Cotton CD bands were characterised for the 1R and 1S systems: the first Cotton bands had values of gCD = −1.20 × 10−3 and +1.23 × 10−3 at 428 nm, the second bands had values of gCD = −1.79 × 10−3 and +1.72 × 10−3 at 403 nm, and the third had values of gCD = −0.26 × 10−3 and +0.26 × 10−3 at 380 nm. The second and third Cotton bands might have arisen from chiral α-phases due to the π–π stacks of the 51- and 52-PF helical chains.8c The first CD band could potentially be assigned to the β-phase even though it was optically active. To distinguish between the new and old β-phases, the new phase is referred to as the chiral β-phase. The chiral β-phase may arise from chiral π–π stacked structures of fully extended anti-coplanar PF (21-helix) chains,8c in which the preferential handedness of the stacks is predominantly determined by the limonene chirality. The previously reported β-phase, though optically inactive, is believed to be highly ordered;8c however, this may be due to an equal amount of left- and right-handed CD-silent, chiral π–π stacks that coexist in solution (conglomerate). The portion of the 428 nm chiral β-phase (2.90 eV), which was responsible for the PL/CPL properties, was determined to be approximately 3.5% from the integral area intensity due to the π–π* transitions ranging from 2.81 eV (441 nm) to 4.13 eV (300 nm).
To verify this conclusion, the UV-vis spectra of PF10-150 K aggregates produced in the chiral tersolvent and achiral cosolvent were analysed (Fig. 1b). When compared to the spectra of the PF10-150 K aggregate produced by limonene chirality transfer, the CD-silent PF10-150 K aggregate dispersed in the mixed chloroform/methanol (1.5/1.5 (v/v)) yielded a very similar broad UV/vis spectrum profile consisting of the 428 nm, 403 nm, and 380 nm bands; however, no detectable CD band emerged in the π–π* transition region. Although PF carrying n-alkyl chains can adopt fully extended anti-coplanar PF chains, chiral π–π stacks of left- and right-handed helicity in an equal probability are partly incorporated in PF aggregates due to slippery fluorene rings. The chirality of the solventlimonene helped to induce a preferential helicity of the stacks.
The CD and UV-vis measurements of PF10-150 K were performed in three limonene/chloroform/alcohol tersolvent systems. Ethanol, isopropyl alcohol (IPA), and 1-octanol were used in place of methanol as the poor polar solvent. As shown in Fig. 2a–2c and Fig. 1a, the CD and UV-vis signal intensities of the PF10-150 K aggregates depend greatly upon the alcohol employed. The absolute magnitudes of the resulting gCD values at the first Cotton CD band around 435 nm in the three tersolvent systems were considerably lower than those of the methanol system. Additionally, when ethanol and 1-octanol were used in place of methanol, the CD signal had the opposite sign, suggesting a switch in the helicity of the stacks.
![CD and UV-vis spectra (a (): 1S, b (⋯): 1R) of CD-active PF10-150 K aggregates formed in three chiral tersolvents at 25 °C. [FL repeating unit] = 2.5 × 10−5 mol L−1 with an 800 rpm (CW) stir rate. (a) limonene/chloroform/ethanol: 0.7/0.3/2.0 (v/v/v), (b) limonene/chloroform/IPA: 0.7/0.3/2.0 (v/v/v), (c) limonene/chloroform/1-octanol: 0.7/0.3/2.0 (v/v/v).](/image/article/2010/PY/b9py00288j/b9py00288j-f2.gif) | ||
| Fig. 2 CD and UV-vis spectra (a (): 1S, b (⋯): 1R) of CD-active PF10-150 K aggregates formed in three chiral tersolvents at 25 °C. [FL repeating unit] = 2.5 × 10−5 mol L−1 with an 800 rpm (CW) stir rate. (a) limonene/chloroform/ethanol: 0.7/0.3/2.0 (v/v/v), (b) limonene/chloroform/IPA: 0.7/0.3/2.0 (v/v/v), (c) limonene/chloroform/1-octanol: 0.7/0.3/2.0 (v/v/v). | ||
For the 1S tersolvent systems, we obtained gCD values of −2.29 × 10−4 at 435 nm for ethanol, −2.49 × 10−4 at 435 nm for 1-octanol, and +1.20 × 10−3 at 428 nm for methanol; the gCD values were undetectable for IPA. The 1R tersolvent systems yielded values having the opposite sign, though with similar absolute values. Among the four achiral poor alcohols tested here, methanol with limonene led to the largest gCD value for the optically active PF10-150 K aggregates. Helicity was preferred with ethanol and 1-octanol, in direct contrast with methanol, when the same handedness of limonene was used. Methanol, the shortest carbon alcohol, may be a specific alcohol.
Optically active PF10-150 K aggregates were also prepared in limonene/good solvent/methanol tersolvent systems, with THF and toluene used as the good solvent in place of chloroform (Fig. S4a–S4b†). Although the spectral characteristics and the signs of the resulting CD and UV-vis spectra in THF and chloroform were almost identical, absolute |gCD| values of 1.11 × 10−3 at 429 nm for THF and 1.21 × 10−3 at 428 nm for chloroform for the 1R and 1S tersolvent systems were measured. However, when toluene was used, the 428 (or 429) nm band in the UV-vis spectra almost disappeared and the absolute |gCD| value at that Cotton band was smaller than 5 × 10−5. Use of the good solvent greatly affected the production of the chiral β-phase. Toluene may have interrupted the effective CH/π interaction between limonene and PF10-150 K and the π/π interaction between the PF10-150 K rings since many potential CH/π interactions between limonene and toluene and between PF10-150 K and toluene are possible.
These chiral π–π, van der Waals, and CH/π forces appear to be weak due to slippery PF rings, leading us to speculate that the induced CD signals were strongly temperature dependent. The CD and UV-vis spectra of the PF10-150 K aggregates prepared at three measurement temperatures, 0 °C, 25 °C, and 50 °C, are shown in Fig. 3, revealing a significant temperature dependence. At higher temperatures, the gCD value and the 428 nm UV-vis band intensity are greatly reduced. The 428 nm CD bands almost disappear at 50 °C as a result of the significant weakening of the π–π stacking force. The preferential helicity of the π–π stacks would likely be abolished if the solution temperature were increased above 50 °C.
![CD and UV-vis spectra (, 1S; ⋯, 1R) of CD-active PF10-150 K aggregates formed in limonene/chloroform/ methanol (0.7/0.3/2.0 (v/v/v)) at (a) 0 °C, (b) 25 °C, and (c) 50 °C with stirring at 800 rpm (CW). [FL repeating unit] = 2.5 × 10−5 mol L−1.](/image/article/2010/PY/b9py00288j/b9py00288j-f3.gif) | ||
| Fig. 3 CD and UV-vis spectra (, 1S; ⋯, 1R) of CD-active PF10-150 K aggregates formed in limonene/chloroform/ methanol (0.7/0.3/2.0 (v/v/v)) at (a) 0 °C, (b) 25 °C, and (c) 50 °C with stirring at 800 rpm (CW). [FL repeating unit] = 2.5 × 10−5 mol L−1. | ||
To test the solvent chirality transfer using other nonpolar terpenes with mirror image forms, bicyclic α-pinenes were compared with the monocyclic limonenes. When using α-pinene, the Cotton CD band due to the PF10-150 K aggregates and induced by the α-pinene chirality was also observed in the region of the π–π* transitions (Fig. 4). However, the absolute gCD magnitudes at the three λext values when using the α-pinenes were one third of those obtained when using the limonenes. These results indicate that the presence of bulky α-pinene in the PF10-150 K aggregates may have weakened the π–π stacking force, leading to a slight loss of helicity of the π–π stacks in addition to the solution temperature effect. The degree of bulkiness and the monocyclic or bicyclic structure greatly affected the formation of the chiral π–π stacks, as evidenced by the CD amplitude.
![CD/UV-vis spectra of PF10-150 K aggregates formed in (1R)- (⋯) and (1S)- () α-pinene/chloroform/methanol (0.7/0.3/2.0 (v/v/v)) with stirring at 800 rpm (CW) at 25 °C. [FL repeating unit] = 2.5 × 10−5 mol L−1.](/image/article/2010/PY/b9py00288j/b9py00288j-f4.gif) | ||
| Fig. 4 CD/UV-vis spectra of PF10-150 K aggregates formed in (1R)- (⋯) and (1S)- () α-pinene/chloroform/methanol (0.7/0.3/2.0 (v/v/v)) with stirring at 800 rpm (CW) at 25 °C. [FL repeating unit] = 2.5 × 10−5 mol L−1. | ||
These results led us to believe that the chiral π/π, van der Waals, and/or chiral CH/π interactions between non-polar PF10-150 K and non-polar limonene with the aid of the proper combination of a good solvent and poor solvent alcohol might be responsible for the efficient production of CD-active PF10-150 K aggregates. Monocyclic limonenes are very promising chiral solvents. The proper choice of a good solvent and poor solvent alcohol were key to a successful solvent chirality transfer experiment. The selection of an achiral alcohol was also important. In conclusion, chiral limonene transformed achiral PF10 into ambidextrous polymer aggregates whose handedness could be switched with the correct choice of limonene chirality and an achiral alcohol.
Vortex effects
A vortex, which can be generated by mechanically stirring a fluid in a cuvette, was recently reported to provide CD and linear dichroism (LD) spectra that dynamically reflect locally different fluidic situations in several supramolecular systems. For example, rod-shaped nanofibers composed of a supramolecularly polymerised zinc porphyrin bearing a pyridyl group and two carboxylic acidgroups provided clear CD and LDspectra. These results were due to a temporal alignment of the nanofibers along the chiral fluidic flows.15In this study, the LD and CD spectroscopic data of the stirred solutions allowed for a spectroscopic visualisation of the vortex flow. PF10-150 K did not exhibit any optical responses in chloroform/methanol under clockwise (CW) or counterclockwise (CCW) stirring at four different stirring speeds (0, 400, 800 and 1300 rpm) (Fig. S5†). When using chloroform/limonene/methanol, the stirring speed did not have any marked influence on the CD/UV-vis spectra of the PF10-150 K aggregates (Fig. S6†) and the stirring direction only weakly affected the CD/UV-vis spectra. The differences in the gCD values were only ± 3% and chiroptical inversion of the CD band by the vortex effect did not occur. The optically active PF10-150 K helicity by limonene chirality transfer was mechanically stable even in the fluidic condition and even in the presence of the weak vortex induction effect. From achiral, CD-silentPF10-150 K, the mirror image limonenes led to the production of ambidextrous CD-active polymer aggregates whose handedness could by switched throught the selection of limonene chirality.
Aggregate size dependency of CD/UV-vis intensities
We next investigated the effects of the aggregate size on the CD/UV-vis spectra using FOM and membrane filter experiments. The changes in the CD and UV-vis spectra after filtration using a series of different pore sizes (0.1 μm pores by Whatman, 1/2/5/10 μm pores by Millipore) are presented in Fig. S7a–7b.† With smaller pore sizes, the absorbance and Cotton CD signals both decreased dramatically (Fig. 5a). However, the value of the gCD factor did not depend on the pore size (Fig. 5b). Both 1R and 1S produced similar results, indicating that the 428 nm CD band dominantly comes from the short ranged helically oriented π–π stacks incorporated in the PF10-150 K aggregates. Using the half intensity in the CD/UV-vis signals between the unfiltered and filtered samples (Fig. 5a), the mean aggregate size was estimated to be approximately 7–8 μm.![Changes in CD/UV-vis intensities (■, 1S; □, 1R) of PF10-150 K aggregates at 25 °C, with stirring at 800 rpm (CW), as a function of membrane filter pore size. (a) Difference in absorbance (left and right circular polarised light) of UV-vis band at 428 nm as a function of membrane filter pore size. (b) The gCD value at 428 nm as a function of membrane filter pore size. (c) Fluorescence optical micrograph of microsphere due to PF10-150 K (unfiltered) aggregate excited at 365 nm. [FL repeating units] = 2.5 × 10−5 mol L−1 in the form of dispersions in 1S/chloroform/methanol (0.7/0.3/2.0 (v/v/v)).](/image/article/2010/PY/b9py00288j/b9py00288j-f5.gif) | ||
| Fig. 5 Changes in CD/UV-vis intensities (■, 1S; □, 1R) of PF10-150 K aggregates at 25 °C, with stirring at 800 rpm (CW), as a function of membrane filter pore size. (a) Difference in absorbance (left and right circular polarised light) of UV-vis band at 428 nm as a function of membrane filter pore size. (b) The gCD value at 428 nm as a function of membrane filter pore size. (c) Fluorescence optical micrograph of microsphere due to PF10-150 K (unfiltered) aggregate excited at 365 nm. [FL repeating units] = 2.5 × 10−5 mol L−1 in the form of dispersions in 1S/chloroform/methanol (0.7/0.3/2.0 (v/v/v)). | ||
To directly characterise the aggregate size, we observed FOM images of PF10-150 K. Fig. 5c shows the fluorescence image excited at 365 nm from the PF10-150 K aggregates dispersed in the 1S/chloroform/methanol tersolvent. The size of the aggregate was in the range of 2–10 μm in diameter. When 1R was used in place of 1S, an almost identical image was observed. The sizes revealed by the FOM experiments were almost consistent with the membrane filter experiment. A similar weak aggregate size dependency was observed for CD-active polysilane aggregates produced from CD-silent polysilane.2d In this case, the observed Cotton effect was assumed to arise from the exciton coupling effect of the nearest neighbour chromophores.
Molecular weight and side chain length dependency
Though the absolute magnitude of the gCD value from the PF10-150 K aggregates was independent of the aggregate size, the absolute gCD values greatly depended on the molecular weight of PF10, the n-alkyl chain length of PFs, and the enantiopurity of limonene.The gCD values of the PF10 aggregates as a function of molecular weight (2.50 × 104 ≤ Mw ≤ 4.89 × 105) in the chiral tersolvent are presented in Fig. 6. PF10-150 K had one of the maximum absolute gCD values, suggesting that there is an ideal molecular weight that would yield the largest CD amplitude. This CD signal may arise from a specific chiral higher order π–π stacking structure, such as cholesteric and/or other chiral liquid crystalline structures.16,17
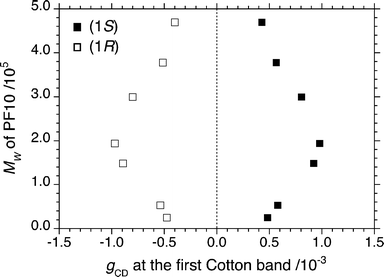 | ||
| Fig. 6 The value of gCD (■, 1S; □, 1R) at 428 nm of PF10 aggregates as a function of molecular weight (Mw) of PF10 in limonene/chloroform/methanol (0.7/0.3/2.0 (v/v/v)) at 25 °C. | ||
Our preliminary limonene chirality transfer experiments using a series of PFs (alkyl; n-hexyl, n-heptyl, n-octyl, n-decyl, n-dodecyl, n-hexadecyl, n-octadecyl), PF10 and poly(9,9-di-n-octylfluorene) (PF8) showed exceptionally intense Cotton CD bands, indicating that the helicity and/or chirality were efficiently induced in these aggregates by van der Waals, CH/π, and/or π–π interactions (Fig. 7). However, other PFs bearing n-alkyl side chains of n-hexyl, n-heptyl, n-dodecyl, n-hexadecyl, and n-octadecyl exhibited very small Cotton CD signals, probably due to weak π–π interactions (Fig. S8†). Therefore, the choice of the proper n-alkyl side chain length is important for the formation of CD-active PF aggregates induced by a poor solvent. Medium lengths of n-alkyl groups efficiently induce chiral π–π stacking structures to yield optically active aggregates.
![CD/UV-vis spectra (, PF10; ⋯, PF8) of PF10-150 K and PF8 (Mw = 0.69 × 105, PDI = 4.2) aggregates produced in limonene/chloroform/methanol (0.7/0.3/2.0 (v/v/v)). [FL repeating unit] = 2.5 × 10−5 mol L−1 with stirring at 800 rpm (CW) at 25 °C.](/image/article/2010/PY/b9py00288j/b9py00288j-f7.gif) | ||
| Fig. 7 CD/UV-vis spectra (, PF10; ⋯, PF8) of PF10-150 K and PF8 (Mw = 0.69 × 105, PDI = 4.2) aggregates produced in limonene/chloroform/methanol (0.7/0.3/2.0 (v/v/v)). [FL repeating unit] = 2.5 × 10−5 mol L−1 with stirring at 800 rpm (CW) at 25 °C. | ||
Circularly polarised luminescence
Fig. 8 shows the first CPL and PL spectra (excited at 380 nm) of the PF10-150 K aggregates produced in the chiral tersolvent at 25 °C. The sign of the CPL signals at 434 nm is identical to that of the 428 nm CD band, which is the longest wavelength of the π–π* CD bands. The absolute magnitude of the gCPL value (∼±2.33 × 10−3) from the CPL band at 434 nm is almost identical to that of the gCD value (∼±1.21 × 10−3) from the CD band at 428 nm. These results suggest that the CPL band at 434 nm was solely derived from the same source as the CD band 428 nm, and the incorporation of a helical higher-ordered π–π stacking structure (the chiral β-phase) in the PF10-150 K aggregates was responsible for these CD and CPL bands. A very small Stokes shift (∼6 nm, 320 cm−1) between the 434 nm CPL and 428 nm CD bands might be due to helical π–π stacks in the aggregates. Although the CD bands came from the ground state of the helical order structure, the CPL signal resulted from the lowest photoexcited energy state in the helical π–π stacking structure.![CPL and PL spectra excited at 380 nm (a (): 1S, b (⋯): 1R) of PF10-150 K aggregates in limonene/chloroform/methanol (0.7/ 0.3/2.0 (v/v/v)). [FL repeating unit] = 5.0 × 10−6 mol L−1 with stirring at 800 rpm (CW) at 25 °C.](/image/article/2010/PY/b9py00288j/b9py00288j-f8.gif) | ||
| Fig. 8 CPL and PL spectra excited at 380 nm (a (): 1S, b (⋯): 1R) of PF10-150 K aggregates in limonene/chloroform/methanol (0.7/ 0.3/2.0 (v/v/v)). [FL repeating unit] = 5.0 × 10−6 mol L−1 with stirring at 800 rpm (CW) at 25 °C. | ||
This conclusion was supported by fluorescence anisotropy experiments of PF10-150 K and its aggregates in the two typical chiral solvent systems for excitation and emission modes under steady-state excitation and detection (Fig. S9a–S9c†). The anisotropy (γ) values of PF10-150 K molecularly dispersed in chloroform remained at approximately 0.4 for the π–π* transitions in the excitation and emission bands (Fig. S9a†). In the case of the PF10-150 K aggregates in the 1S and 1R tersolvents, the γ values were similar (at approximately 0.4) for the π–π* transitions in the excitation and emission bands (Fig. S9b–S9c†), indicating a collinear arrangement of chromophores and fluorophores in the π–π stacks, which are parallel to the main chain axis of PF10-150 K.
Limonene enantiopurity dependency
The amplification of the most significant helix is known as the majority rule. Optically active chain-like polymers and supramolecular π–π stacks with a preferential screw-sense are nonlinearly amplified by the enantiopurity (ee) of chiral pendant groups.16 However, for the optically active PF10-150 K aggregates formed in a mixture of 1R and 1S tersolvent systems, the gCD values changed approximately linearly by switching the enantiopurity of limonene (Fig. 9). A similar enantiopurity dependency of the chiral solvent was observed for the CD-active polysilane aggregates produced from the CD-silent polysilane.2d Because the poor solvent was added at once, nucleation seed and growth occurred quickly, and the right-handed and left-handed aggregates were predominantly generated proportionally to the relative ratio of 1R and 1S. Fig. 10a and 10b compare the CD and CPL spectra of the PF10-150 K aggregates prepared by the normal and reverse modes. In addition, Fig. S10a and S10b compare the circularly unpolarised (UV-vis and PL spectra) spectra of the PF10-150 K aggregates prepared by the normal and reverse modes.† No significant differences in the magnitude or sign between the CD and CPL spectra were observed. Similarly, there were no significant differences in the magnitude, wavelength, or spectral shapes between the UV-vis and PL spectra.![The value of gCD at 428 nm of PF10-150 K aggregates as a function of limonene enantiopurity (ee), produced in limonene/chloroform/methanol (0.7/0.3/2.0 (v/v/v)). [FL repeating unit] = 2.5 × 10−5 mol L−1 with stirring at 800 rpm (CW) at 25 °C.](/image/article/2010/PY/b9py00288j/b9py00288j-f9.gif) | ||
| Fig. 9 The value of gCD at 428 nm of PF10-150 K aggregates as a function of limonene enantiopurity (ee), produced in limonene/chloroform/methanol (0.7/0.3/2.0 (v/v/v)). [FL repeating unit] = 2.5 × 10−5 mol L−1 with stirring at 800 rpm (CW) at 25 °C. | ||
![CD and CPL spectra (a (): 1S, b (⋯): 1R) of PF10-150 K aggregates produced in limonene/chloroform/methanol (0.7/0.3/2.0 (v/v/v)) at 800 rpm (CW) and 25 °C. (a) Addition order of first limonene and then methanol, (b) addition order of first methanol and then limonene. For the CD measurement, [FL repeating unit] = 2.5 × 10−5 mol L−1 and for the CPL measurement, [FL repeating unit] = 5.0 × 10−6 mol L−1.](/image/article/2010/PY/b9py00288j/b9py00288j-f10.gif) | ||
| Fig. 10 CD and CPL spectra (a (): 1S, b (⋯): 1R) of PF10-150 K aggregates produced in limonene/chloroform/methanol (0.7/0.3/2.0 (v/v/v)) at 800 rpm (CW) and 25 °C. (a) Addition order of first limonene and then methanol, (b) addition order of first methanol and then limonene. For the CD measurement, [FL repeating unit] = 2.5 × 10−5 mol L−1 and for the CPL measurement, [FL repeating unit] = 5.0 × 10−6 mol L−1. | ||
Therefore, regardless of the addition order of limonene (normal or reverse), the optical activity of the PF10-150 K stacks in aggregates could easily switch between the left- and right-handed preferences by the limonene chirality due to the very slippery nature of the PF10 stacks. When limonene was absent or in a racemic mixture, the left- and right-handed PF10 stacks coexisted as conglomerates in solution, leading to CD-silentPF stacks.
π–π Stacks revealed by wide-angle X-ray scattering study
The basis for the differences in the stacking structure from the PF10 aggregates with or without limonene and the locations of the limonene molecules in the optically active PF10 aggregates remain unknown. Recently, small-angle X-ray scattering experiments have revealed that poly(9,9-di-n-alkylfluorene) derivatives can self-organise into π–π stacking structures with the help of a poor solvent.17 Therefore, to investigate the structures of the PF10 aggregates, we carried out WAXD experiments. The WAXD data for the PF10 aggregates prepared in the 1R and 1S tersolvent without limonene are shown in Fig. 11b and 11c.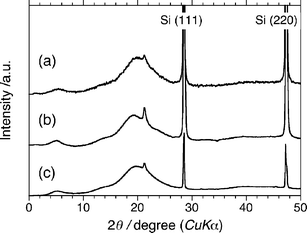 | ||
| Fig. 11 WAXD of PF10 (Mw = 1.9 × 105, PDI = 5.1) cast from solution of (a) chloroform, (b) 1S/chloroform/methanol (0.7/0.3/2.0 (v/v/v)), and (c) 1R/chloroform/methanol (0.7/0.3/2.0 (v/v/v)). | ||
Without limonene (Fig. 11a), there are two major scattering peaks, which correspond to d-spacings of 16.3 Å (2θ = 5.4°) and 4.5 Å (2θ = 19.8°). The peaks are indicative of local (i.e., short-range) biaxiality in the stacking of the PF10 molecules. The d-spacings of 16.3 Å and 4.5 Å were assigned to the interchain distance of the PF10 chains with a spacer of decyl chains and the van der Waals distance of the π–π stacks, respectively (Fig. 12, top). Unlike the more conventional hairy rod polymers, where the side chains emanate in a roughly coplanar manner from the aromatic backbone, the side chains in PF10 are perpendicular to the aromatic plane. Therefore, it was expected that the value of 16.3 Å would be much shorter than the sum of the two decyl chain lengths, which is estimated to be 25 Å, on the assumption that two decyl chains adopt the extended form normal to the fluorene rings. The observed d-spacing of 16.3 Å suggests that the n-decyl chains are significantly tilted relative to the direction of the edge-to-edge stacking of the PF10 main chains (Fig. 12, top). The intense scattering of 4.1 Å (2θ = 21.4°) was assigned to the interchain distance of the nearest neighbouring n-decyl chains in the direction of the polymer backbone.
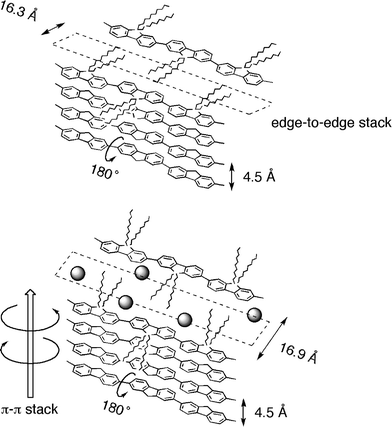 | ||
| Fig. 12 Proposed π–π stacking models of PF10 aggregates revealed by WAXD and CD/CPL experimental results. | ||
For the 1R system, the d-spacings of ∼4.5 Å (2θ = 19.6°) due to the π–π stacks and of ∼4.1 Å (2θ = 21.4°) due to the interchain distance between the side chains are the same as those of the PF10 aggregates without limonene. However, the value of 16.9 Å (2θ = 5.2°) is 0.6 Å longer than the value (16.3 Å) from the PF10 aggregates without limonene. The three peaks from 1S are identical to those from 1R, suggesting that the n-decyl chains are raised in the direction of the edge-to-edge stacking of the PF10 main chains due to the presence of limonene. Therefore, the limonene molecules likely exist closer to the n-decyl chains, leading to the incorporation of limonene in PF10 aggregates, regardless of the limonene chirality (Fig. 12, bottom).
Conclusions
Optically active PF aggregates in limonene/good solvent/poor solvent systems were generated when CD-silent polyfluorenes bearing n-decyl and n-octyl side chains (as the hostpolymers) and limonene (as the guest molecular solvent) were used. The aggregates exhibited intense Cotton CD and CPL bands in the UV-vis region, depending on the molecular weights and chain length of the side chain of PF, the chiral solvent enantiopurity, the chemical structure of the chiral solvent, and the temperature of the tersolvent. The portion of the π–π stacks responsible for the CPL/PL bands was estimated to be 3.5% at 25 °C, which results from the inherently slippery π–π stacking force.The terpene chirality transfer approach allows for the rapid production of CPL-/CD-functionalised polymer aggregates from CD-silentPF under mild conditions and at ambient temperature. Due to the efficiency of the precipitation method, the product yield can be almost 100%. The monocyclic terpenes volatile 1S and 1R (bp 176 °C), which have mint/pine and orange/lemon aromas, respectively, are the most promising chiral solvents for producing ambidextrous chiroptical polymers since they are inexpensive and renewable.2j The resulting luminous polymer aggregates are promising as chiroptical inks for circular polarisation-related device applications.
The understanding provided herein of the solvent chirality transfer experiment is applicable to the production of various optically active polymeric fluorophores that are able to emit not only blue colour but also green and red light when using CD-silent artificial polymers and the proper selection of chiral and achiral solvents.
Acknowledgements
We are grateful for the financial support received from the Initiatives for Attractive Education in Graduate Schools (FY2007), the Foundation for Nara Institute of Science and Technology (FY2009), a Grant-in-Aid for Science Research from JSPS (21655041, FY2009–FY2010) and the SEKISUI Chemical Grant Program for Research on Manufacturing Based on Learning from Nature (FY2009). We thank Drs Hisao Yanagi, Yasuchika Hasegawa, Atsushi Ikeda, Hironari Kamikubo, Masanobu Naito, Kotohiro Nomura, Hisanari Onouchi, Wei Zhang, and Masaaki Ishikawa for their stimulating discussions and helpful comments. We thank Fumiko Ichiyanagi for technical guidance in the CD/CPL/UV-vis/FL, GPC, and GC measurements. We also thank Satoshi Fukao, Jalliah Bindi ABD Jalil, Yoshifumi Kawagoe, Yasuko Nakamura, and Takashi Mori for fruitful discussions throughout these experiments.References
- For reviews and books, see (a) G. Wald, Ann. N. Y. Acad. Sci., 1957, 69, 352–368 CrossRef CAS; (b) W. Thiemann and W. Darge, Origins Life, 1974, 5, 263–283 CrossRef CAS; (c) S. F. Mason, Nature, 1984, 311, 19–23 CAS; (d) V. I. Goldanskii and V. V. Kuz'min, Sov. Phys. Usp., 1989, 32, 1–28 CrossRef; (e) W. A. Bonner, Top. Stereochem., 1988, 18, 1–96 Search PubMed; (f) L. Orgel, Nature, 1992, 358, 203–209 CrossRef CAS; (g) Advances in Biochemistry, ed. G. Pályi, C. Zucchi and L. Caglioti, Elsevier, Amsterdam, 1999 Search PubMed; (h) B. L. Feringa and R. A. van Delden, Angew. Chem., Int. Ed., 1999, 38, 3418–3438 CrossRef; (i) Chirality in Natural and Applied Science, ed. L. W. Lough and I. W. Wainer, Blackwell, Oxford, 2002 Search PubMed; (j) M. Quack, Angew. Chem., Int. Ed., 2002, 41, 4618–4630 CrossRef CAS; (k) K. Soai and T. Kawasaki, Top. Curr. Chem., 2008, 284, 1–33 CAS; (l) A. Guijarro, M. Yus, The Origin of Chirality in The Molecules of Life, RSC, London, 2009 Search PubMed.
- (a) M. M. Green, N. C. Peterson, T. Sato, A. Teramoto, R. Cook and S. Lifson, Science, 1995, 268, 1860–1866 CrossRef CAS; (b) E. Peeters, M. P. T. Christiaans, R. A. J. Janssen, H. F. M. Schoo, H. P. J. M. Dekkers and E. W. Meijer, J. Am. Chem. Soc., 1997, 119, 9909–9910 CrossRef CAS; (c) M. Fujiki, J. Am. Chem. Soc., 2000, 122, 3336–3343 CrossRef CAS; (d) H. Nakashima, J. R. Koe, K. Torimitsu and M. Fujiki, J. Am. Chem. Soc., 2001, 123, 4847–4848 CrossRef CAS; (e) H. Nakako, R. Nomura and T. Masuda, Macromolecules, 2001, 34, 1496–1502 CrossRef CAS; (f) A. P. H. J. Schenning, P. Jonkheijm, E. Peeters and E. W. Meijer, J. Am. Chem. Soc., 2001, 123, 409–416 CrossRef CAS; (g) T. Aoki, T. Kaneko, N. Maruyama, A. Sumi, M. Takahashi, T. Sato and M. Teraguchi, J. Am. Chem. Soc., 2003, 125, 6346–6347 CrossRef CAS; (h) J. Tabei, R. Nomura, F. Sanda and T. Masuda, Macromolecules, 2003, 36, 8603–8608 CrossRef CAS; (i) A. Saxena, G. Guo, M. Fujiki, Y. Yang, A. Ohira, K. Okoshi and M. Naito, Macromolecules, 2004, 37, 3081–3083 CrossRef CAS; (j) H.-Z. Tang, B. M. Novak, J. He and P. L. Polavarapu, Angew. Chem., Int. Ed., 2005, 44, 7298–7301 CrossRef CAS; (k) G. Lakhwani, S. C. J. Meskers and R. A. J. Janssen, J. Phys. Chem. B, 2007, 111, 5124–5131 CrossRef CAS; (l) A. M. Buono, I. Immediate, P. Rizzo and G. Guerra, J. Am. Chem. Soc., 2007, 129, 10992–10993 CrossRef CAS; (m) J. Luijten, E. J. Vorenkamp and A. J. Schouten, Langmuir, 2007, 23, 10772–10778 CrossRef CAS; (n) T. Fujii, M. Shiotsuki, Y. Inai, F. Sanda and T. Masuda, Macromolecules, 2007, 40, 7079–7088 CrossRef CAS; (o) G. Cipparrone, P. Pagliusi, C. Provenzano and V. P. Shibaev, Macromolecules, 2008, 41, 5992–5996 CrossRef CAS; (p) E. Yashima, K. Maeda and Y. Furusho, Acc. Chem. Res., 2008, 41, 1166–1180 CrossRef CAS.
- (a) A. Satrijo, S. C. J. Meskers and T. M. Swager, J. Am. Chem. Soc., 2006, 128, 9030–9031 CrossRef CAS; (b) G. A. Hembury, V. V. Borovkov and Y. Inoue, Chem. Rev., 2008, 108, 1–73 CrossRef CAS; (c) H. Matsukizono, K. Kuroiwa and N. Kimizuka, J. Am. Chem. Soc., 2008, 130, 5622–5623 CrossRef CAS; (d) Chirality at the Nanoscale: Nanoparticles, Surfaces, Materials and More, ed. D. B. Amabilino, Wiley-VCH, Weiheim, 2009 Search PubMed.
- (a) S. H. Chen, D. Katsis, A. W. Schmid, J. C. Mastrangelo, T. Tsutsui and T. N. Blanton, Nature, 1999, 397, 506–508 CrossRef CAS; (b) H. P. Chen, D. Katsis, J. C. Mastrangelo, S. H. Chen, S. D. Jacobs and P. J. Hood, Adv. Mater., 2000, 12, 1283–1286 CrossRef CAS; (c) R. Ozaki, Y. Matsuhisa, M. Ozaki and K. Yoshino, Appl. Phys. Lett., 2004, 84, 1844–1846 CrossRef CAS; (d) S. Furumi and Y. Sakka, Adv. Mater., 2006, 18, 775–780 CrossRef CAS; (e) N. Y. Ha, Y. Ohtsuka, S. M. Jeong, S. Nishimura, G. Suzaki, Y. Takanishi, Y. Ishikawa and H. Takezoe, Nat. Mater., 2008, 7, 43–47 CrossRef CAS.
- (a) R. A. van Delden, N. P. M. Huck, J. J. Piet, J. M. Warman, S. C. J. Meskers, H. P. J. M. Dekkers and B. L. Feringa, J. Am. Chem. Soc., 2003, 125, 15659–15665 CrossRef CAS; (b) F. C. Spano, S. C. J. Meskers, E. Hennebicq and D. Beljonne, J. Am. Chem. Soc., 2007, 129, 7044–7054 CrossRef CAS; (c) S. Petoud, G. Muller, E. G. Moore, J. Xu, J. Sokolnicki, J. P. Riehl, U. N. Le, S. M. Cohen and K. N. Raymond, J. Am. Chem. Soc., 2007, 129, 77–83 CrossRef CAS; (d) K. Do, F. C. Muller and G. Muller, J. Phys. Chem. A, 2008, 112, 6789–6793 CrossRef CAS; (e) J. L. Lunkley, D. Shirotani, K. Yamanari, S. Kaizaki and G. Muller, J. Am. Chem. Soc., 2008, 130, 13814–13815 CrossRef CAS.
- (a) H. Yao, K. Miki, N. Nishida, A. Sasaki and K. Kimura, J. Am. Chem. Soc., 2005, 127, 15536–15543 CrossRef CAS; (b) Y. Imai, K. Kawano, Y. Nakano, K. Kawaguchi, T. Harada, T. Sato, M. Fujiki, R. Kuroda and Y. Matsubara, New J. Chem., 2008, 32, 1110–1112 RSC; (c) Y. Imai, K. Murata, N. Asano, Y. Nakano, K. Kawaguchi, T. Harada, T. Sato, M. Fujiki, R. Kuroda and Y. Matsubara, Cryst. Growth Des., 2008, 8, 3376–3379 CrossRef CAS; (d) S. D. Elliott, M. P. Moloney and Y. K. Gun'ko, Nano Lett., 2008, 8, 2452–2457 CrossRef CAS.
- For reviews, see (a) M. J. Leclerc, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2867–2873 CrossRef CAS; (b) A. C. Grimsdale and K. Müllen, Adv. Polym. Sci., 2006, 199, 1–82 CAS; (c) U. Scherf and D. Neher, Adv. Polym. Sci., 2008, 212 Search PubMed.
- (a) Q. Pei and Y. Yang, J. Am. Chem. Soc., 1996, 118, 7416–7417 CrossRef CAS; (b) M. Grell, D. D. C. Bradley, M. Inbasekarun and E. P. Woo, Adv. Mater., 1997, 9, 798–802 CrossRef CAS; (c) M. Grell, D. D. C. Bradley, G. Ungar, J. Hill and K. S. Whitehead, Macromolecules, 1999, 32, 5810–5817 CrossRef CAS; (d) M. Oda, S. C. J. Meskers, H. G. Nothofer, U. Scherf and D. Neher, Synth. Met., 2000, 111–112, 575–577 CrossRef CAS; (e) M. Oda, H. G. Nothofer, G. Lieser, U. Scherf, S. C. J. Meskers and D. Neher, Adv. Mater., 2000, 12, 362–365 CrossRef CAS; (f) G. Lieser, M. Oda, T. Miteva, A. Meisel, H. G. Nothofer, U. Scherf and D. Neher, Macromolecules, 2000, 33, 4490–4495 CrossRef CAS; (g) H.-Z. Tang, M. Fujiki and M. Motonaga, Polymer, 2002, 43, 6213–6220 CrossRef CAS; (h) H.-Z. Tang, M. Fujiki and T. Sato, Macromolecules, 2002, 35, 6439–6445 CrossRef CAS; (i) A. C. Grimsdale and K. Müllen, Adv. Polym. Sci., 2006, 199, 1–82 CAS; (j) Y. Liu, T. Murao, Y. Nakano, M. Naito and M. Fujiki, Soft Matter, 2008, 4, 2396–2401 RSC; (k) G. Lakhwani and S. C. J. Meskers, Macromolecules, 2009, 42, 4220–4223 CrossRef CAS.
- For books and reviews, see (a) M. Nishio, M. Hirota and Y. Umezawa, The CH/π Interaction: Evidence, Nature, and Consequences; Wiley–VCH: New York, 1998 Search PubMed; (b) G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond: In Structural Chemistry and Biology; Oxford University Press: Oxford, 2001 Search PubMed; (c) P. Hobza and Z. Havlas, Chem. Rev., 2000, 100, 4253–4264 CrossRef CAS; (d) M. Fujiki and A. Saxena, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4637–4650 CrossRef CAS.
- (a) E. Yashima, T. Matsushima and Y. Okamoto, J. Am. Chem. Soc., 1997, 119, 6345–6359 CrossRef CAS; (b) E. Yashima, K. Maeda and T. Yamanaka, J. Am. Chem. Soc., 2000, 122, 7813–7814 CrossRef CAS; (c) K. Maeda, K. Morino, Y. Okamoto, T. Sato and E. Yashima, J. Am. Chem. Soc., 2004, 126, 4329–4342 CrossRef CAS; (d) K. Maeda and E. Yashima, Top. Curr. Chem., 2006, 265, 47–88 CAS; (e) Y. Hase, K. Nagai, H. Iida, K. Maeda, N. Ochi, K. Sawabe, K. Sakajiri, K. Okoshi and E. Yashima, J. Am. Chem. Soc., 2009, 131, 10719–10732 CrossRef CAS.
- (a) J. J. L. M. Cornelissen, A. E. Rowan, R. J. M. Nolte and N. A. J. M. Sommerdijk, Chem. Rev., 2001, 101, 4039–4070 CrossRef CAS; (b) L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4098 CrossRef CAS; (c) E. Yashima, K. Maeda and T. Nishimura, Chem.–Eur. J., 2004, 10, 42–51 CrossRef CAS.
- (a) B. J. Bosnich, J. Am. Chem. Soc., 1967, 89, 6143–6148 CrossRef CAS; (b) M. M. Green, C. Khatri and N. C. Peterson, J. Am. Chem. Soc., 1993, 115, 4941–4942 CrossRef CAS; (c) C. A. Khatri, Y. Pavlova, M. M. Green and H. Morawetz, J. Am. Chem. Soc., 1997, 119, 6991–6995 CrossRef.
- D. F. Eaton, Pure Appl. Chem., 1988, 60, 1107–1114 CrossRef CAS.
- H. P. J. M. DekkersIn Circular Dichroism: Principles and Applications, ed. N. Berova, K. Nakanishi, R. W. Woody, Wiley–VCH, New York, 2nd edn, 2000, ch. 7 Search PubMed.
- (a) A. Tsuda, M. A. Alam, T. Harada, T. Yamaguchi, N. Ishii and T. Aida, Angew. Chem., Int. Ed., 2007, 46, 8198–8202 CrossRef CAS; (b) M. Wolffs, S. J. George, Z. Tomovic, S. C. J. Meskers, A. P. H. J. Schenning and E. W. Meijer, Angew. Chem., Int. Ed., 2007, 46, 8203–8205 CrossRef CAS.
- (a) M. Green, N. C. Peterson, T. Sato, A. Teramoto, R. Cook and S. Lifson, Science, 1995, 268, 1860–1866 CrossRef CAS; (b) J. van Gestel, A. R. A. Palmans, B. Titulaer, J. A. J. M. Vekemans and E. W. Meijer, J. Am. Chem. Soc., 2005, 127, 5490–5494 CrossRef CAS.
- (a) M. Knaapila, V. M. Garamus, F. B. Dias, L. Almásy, F. Galbrecht, A. Charas, J. Morgado, H. D. Burrows, U. Scherf and A. P. Monkman, Macromolecules, 2006, 39, 6505–6512 CrossRef CAS; (b) M. Knaapila, F. B. Dias, V. M. Garamus, L. Almásy, M. Torkkeli, K. Leppänen, F. Galbrecht, E. Preis, H. D. Burrows, U. Scherf and A. P. Monkman, Macromolecules, 2007, 40, 9398–9405 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary experimental details, CD spectra in other solvents and other PF derivatives, vortex effects, fluorescence and fluorescence anisotropy of PF10, and preparation conditions of PF10 aggregates. See DOI: 10.1039/b9py00288j |
| This journal is © The Royal Society of Chemistry 2010 |