Detailed investigation of the propagation rate of urethane acrylates
Christopher
Barner-Kowollik
*a,
Francesca
Bennet
a,
Maria
Schneider-Baumann
a,
Dominik
Voll
a,
Thomas
Rölle
b,
Thomas
Fäcke
b,
Marc-Stephan
Weiser
b,
Friedrich-Karl
Bruder
b and
Tanja
Junkers
a
aPreparative Macromolecular Chemistry, Institut für Technische Chemie und Polymerchemie, Karlsruhe Institute of Technology (KIT), Engesserstr. 18, 76128, Karlsruhe, Germany. E-mail: christopher.barner-kowollik@kit.edu; Web: www.macroarc.de Fax: +49 721 608 5740; Tel: +49 721 608 5641
bBayer MaterialScience AG, 51368, Leverkusen, Germany
First published on 19th January 2010
Abstract
Temperature dependent propagation rate coefficients, kp, are determined for four acrylate monomers containing a carbamate moiety via the pulsed laser polymerization-size exclusion chromatography (PLP-SEC) technique. Therefore, the Mark–Houwink–Kuhn–Sakurada coefficients K and a of the respective polymers were additionally determined via triple-detection SEC. The monomers under investigation were synthesized from hydroxyethyl acrylate, hydroxyl(iso)propyl acrylate as well as phenyl isocyanate and hexyl isocyanate, respectively, in all four possible combinations. For 2-(phenylcarbamoyloxy)ethyl acrylate (PhCEA) an activation energy of 14.3 kJ mol−1 and a frequency factor of A = 1.2 × 107 L·mol−1 s−1 are obtained for kp. The MHKS parameters for poly(PhCEA) are K = 8.3 × 10−5 dL g−1 and a = 0.677. For 2-(phenylcarbamoyloxy)isopropyl acrylate (PhCPA) an activation energy of 14.2 kJ mol−1 and a frequency factor of A = 4.9 × 106 L mol−1 s−1 are found for kp and the MHKS parameters for poly(PhCPA) read K = 10.3 × 10−5 dL g−1 and a = 0.657. The activation parameters of kp of 2-(hexylcarbamoyloxy)ethyl acrylate (HCEA) are EA = 13.3 kJ mol−1 and A = 6.6 × 106 L mol−1 s−1 with K = 36.0 × 10−5 dL g−1 and a = 0.552 for poly(HCEA). For 2-(hexylcarbamoyloxy)isopropyl acrylate (HCPA) EA is 14.1 kJ mol−1 and A = 6.6 × 106 L mol−1 s−1 with K = 26.0 × 10−5 dL g−1 and a = 0.587 for poly(HCPA). All rate measurements were performed in 1 M solutions in butyl acetate. The fast propagating nature and reduced activation energy of the monomers may be understood on the basis of the increased nucleophilicity that is induced by the carbamate functionality present in all monomers. Rate-increasing effects from solvent polarity and/or from H-bonding can, however, not be excluded and might also contribute to the observed high propagation rates.
Introduction
Pulsed laser polymerization (PLP) coupled with size exclusion chromatography (SEC)1–3 is today the standard technique for the determination of propagation rate coefficients, kp. It has been applied to a large number of monomers4 and benchmark values for a number of commonly used monomers have been published by an IUPAC working party.5–10 The success of the PLP-SEC technique relies mostly on the high accuracy of the method as the uncertainty of the resulting coefficients is for most parts only given by the error in the SEC determination of molecular weight distributions. Even the problem of SEC accuracy may soon be overcome by employing absolute methods such as coupled SEC-ESI-MS methodologies.11 While accurate rate coefficients are available, progress in understanding the influences that determine the actual propagation rate is steady, yet slow. Vinylic monomers are usually categorized into families with distinct reactivity such as acrylates, methacrylates, styrenics or other classes. The differences in reactivity can often be directly correlated to the general reactivity of the vinylic centre. However, substituents play a significant role and also affect the reaction rate, and similar trends with size and structure of the ester side chain within the acrylate or methacrylate family can be observed. Today it is widely accepted that the rate of propagation is not only determined by steric factors, but also largely by dipolar interactions, be it intermolecular interactions with the reaction media,12 by intramolecular shielding of dipoles that affect the transition state of the reaction13 or H-bonding.14 For example, for alkyl acrylates, kp increases when the size of the ester is increased, a result that may not be expected from a steric point of view. An explanation for this result can, however, be given by the increased shielding of the dipolar interaction between two ester moieties from the larger alkyl chain, thus allowing for a decreased barrier for rotation in the transition state that allows the reaction to proceed faster.13Recently, Bowman and co-workers published a series of papers noting the particularly rapid polymerization in UV curing experiments of (meth)acrylates carrying a carbamate moiety on the ester side chain.15–20 These authors proposed different theories for the reason why the polymerization occurs rapidly. These findings are of industrial interest, since urethane acrylates are today established as active ingredients of a whole set of radiation curing coatings, adhesives and sealants.21 However, a clear differentiation between individual effects besides observing overall polymerization rates for radiation curing materials is difficult to achieve and not well established. The overall polymerization rate in complex systems and in single molecules is affected by not only the individual rate of propagation, but also highly dependent on the diffusion controlled termination rate, and—as is often the case with acrylate monomers—transfer reactions. A clear answer as to how the carbamate moieties affect the reactivity of the acrylates could thus not be provided. Therefore, we decided to apply high-frequency pulsed laser polymerization (PLP) (which allows for the determination of kp of acrylates in a broad temperature regime)22–24 to a series of carbamate containing acrylates to elucidate what effect the secondary functionalities have on the rate of propagation. Thus, we wish to explore whether the reactivity of selected urethane acrylates is dominated by the nature of the propagation reaction or by alternative factors. Therefore, a representative set of monomers as depicted in Scheme 1 were under investigation.
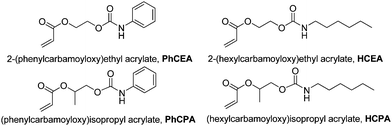 | ||
| Scheme 1 Monomers under investigation in this work. | ||
In previous studies,152-(phenylcarbamoyloxy)ethyl acrylate, PhCEA, was shown to be a particularly fast polymerizing monomer, thus the present investigations were based on the monomer PhCEA. To establish which functional group in the monomer is responsible for its high reactivity, derivatives of PhCEA were also synthesized. Analogues of monomers with either a more flexible hexyl side chain and/or an isopropyl bridge between the ester and carbamate function were selected. The long alkyl group was chosen as a structural element in comparison to the phenyl group in order to test what effect a more flexible side chain may have on the rate of propagation. With alkyl acrylates, the rate increases with the size of the side chain, an effect also seen in alkyl methacrylates,4 which is frequently rationalized by a shielding of the dipole of the ester from the longer alkyl chain. Such shielding results in a more favourable transition state structure that effectively results in larger pre-exponential factors in the Arrhenius equation.13 In addition, because carbamates can form quite strong H-bonding, it may be hypothesized that the phenyl group allows for an easier approach of monomers to the reactive centre as well as some kind of pre-organization of monomers in solution. In fact, PhCEA as well as its analogues show a strong tendency to form crystals due to high crystallization enthalpies. The variation of introducing an isopropyl bridge between the two key functionalities followed that line of thought. The additional methyl group results in a less flexible orientation of the carbamate moiety, which should have a pronounced effect on the sterics of the reaction. In addition, Bowman and colleagues advanced the theory that one of the reasons for the rapid polymerization might be a potential transfer reaction of the active radical centre onto the ethyl bridge. This hypothesis was supported by the observation of a decrease in the reaction rate when the ethyl bridge was methylated. While such an explanation is rather hypothetical (as it requires a significant driving force for the transfer step to occur, due to the largely increased reactivity of the generated side-chain radical), it can be tested with ease viaPLP, as PLP-SEC experiments are only successful in cases where transfer to polymer reactions do not interfere.9,25–27 If too many transfer steps take place between two consecutive laser pulses, the chain length–time correlation is lost and no characteristic structure in the molecular weight distribution will be observable in the generated polymer. Should a transfer reaction as postulated take place, it should also occur intermolecularly (as well as onto the monomer itself), thus resulting in the failure of the PLP experiment.
It should be noted that the two monomers, PhCPA and HCPA, consist of a mixture of isomers as they were synthesized from commercial hydroxypropyl acrylate, which is an approximately 2 : 1 mixture of isomers as shown in Scheme 2.
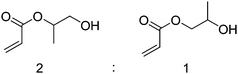 | ||
| Scheme 2 Mixture of isomers in the starting material for PhCPA and HCPA synthesis based on NMR integration. | ||
PLP-SEC requires the accurate determination of molecular weight distributions. SEC is usually the method of choice due to its highly accurate polymer concentration detection viarefractive index detectors. However, the SEC technique must be used with care. The determination of molecular weight is limited in its accuracy, especially when universal calibration is employed. In fact, the systematic error from SEC analysis by far exceeds the inherent error from PLP, thus increasing the importance of correct data handling. In acrylatepolymerizations side reactions occur, analogous to ethyleneradical polymerization,28 that lead to an extensive formation of branching points in the polymer. Two kinds of branching must be distinguished:25 one is short-chain branching that is formed upon the so-called backbiting reaction and the other is long-chain branching, formed upon random inter- or intramolecular transfer. Recently, it was shown that short-chain branching is uncomplicated with respect to SEC analysis because short-chain branches are equally distributed over the chains and do not significantly change the intrinsic viscosity.29 On the other hand, long-chain branches have a pronounced effect on viscosity and at least in a certain molecular weight window the SEC analysis of a polyacrylate sample can yield erroneous apparent molecular weights as the Mark–Houwink–Kuhn–Sakurada (MHKS) relation does not hold.29 Material produced viaPLP is rather inhomogeneous with regard to chain branching because the laser pulse action interrupts the equilibration between mid-chain and end-chain radicals. As a consequence, periodic levels of chain branching can be detected in such samples.30
Nevertheless, for reasons given below (and having been recently outlined)24 we have chosen to analyze the polyacrylates under investigation in the current study by means of universal calibration. Before presenting the results of the PLP experiments and the discussion of the derived rate coefficients, we will focus on the determination of MHKS parameters for the given monomers, which values themselves may also be of high interest.
Experimental
Materials
2-Hydroxyethyl acrylate (Alfa Aesar, 95%), hydroxypropyl acrylate (Aldrich, 95%, mixture of isomers), phenyl isocyanate (Fluka, ≥99%), hexyl isocyanate (Acros, 99%), dibutyl tin laurate (DBTL) (Alfa Aesar, 95%), ethyl acetate (Fischer Scientific, AR grade), butyl acetate (Aldrich, 99.7%), n-octanethiol (Acros Organics, 97%) and 2,2-dimethoxy phenylacetophenone (DMPA, Aldrich) were all used as received. 2,2′-Azobisisobutyronitrile (AIBN) was recrystallized twice from ethanol. Tetrahydrofuran (THF) (Scharlau, HPLC grade) was used as received.Monomer synthesis
The synthesis procedure for PhCEA is given. All other monomers were synthesized accordingly: 50 g (0.42 mol) of phenyl isocyanate and 100 mg hydroquinone are dissolved in 50 mL ethyl acetate and one drop dibutyl tinlaurate is added as catalyst. The solution is heated to 60 °C and 48.74 g (0.42 mol, 1 eq) 2-hydroxyethyl acrylate is added dropwise over a period of 1 h. The mixture is then stirred at constant temperature for further 14 h. The solution is passed over a column of basic alumina to remove the inhibitor and subsequently, the solvent is removed in vacuo. A white solid is obtained in quantitative yield that is recrystallized from ethyl acetate. Note: in PhCPA and HCPA, because of the isomeric mixture of hydroxypropyl acrylate used in monomer synthesis, some peaks in NMR appear twice at very similar chemical shifts. PhCEA: 1H NMR (CDCl3) δ (ppm): 4.42 (s, 4H), 5.74 (d, 1H), 6.16 (dd, 1H), 6.46 (d, 1H), 6.80 (s, 1H), 7.06 (m, 1H), 7.28 (m, 4H); 13C NMR (CDCl3) δ (ppm): 62.4, 62.8, 118.6, 127.9, 129.0, 131.4, 137.5, 165.9, 171.1. Elemental analysis: found: C, 61.3; H, 5.6; N, 5.8. C12H12NO4 requires: C, 61.5; H, 5.2; N, 6.0%. PhCPA: 1H NMR (CDCl3) δ (ppm): 1.25 (m, 3H), 4.18 (m, 2H), 5.12 (m, 1H), 5.76 (d, 1H), 6.06 (dd, 1H), 6.35 (d, 1H), 6.74 (s, 1H), 6.99 (m, 1H), 7.26 (m, 4H); 13C NMR (CDCl3) δ (ppm): 16.9, 66.4, 67.0, 68.9, 69.4, 118.8, 123.7, 128.1, 129.2, 131.7, 138.0, 153.0, 166.1%. Elemental analysis: found: C, 62.6; H, 6.3; N, 5.5. C13H14NO4 requires: C, 62.9; H, 5.7; N, 5.6%. HCEA: 1H NMR (CDCl3) δ (ppm): 0.81 (m, 3H), 1.22 (m, 4H), 1.42 (m, 2H), 3.10 (m, 2H), 4.26 (m, 4H), 4.78 (s, 1H), 5.79 (d, 1H), 6.08 (dd, 1H), 6.37 (d, 1H); 13C NMR (CDCl3) δ (ppm): 14.2, 23.0, 26.5, 30.0, 31.6, 41.2, 62.8, 128.2, 131.4, 156.2, 166.2. Elemental analysis: found: C, 60.1; H, 8.8; N, 5.6. C12H20NO4 requires: C, 59.5; H, 8.3; N, 5.8%. HCPA: 1H NMR (CDCl3) δ (ppm): 0.81 (m, 3H), 1.22 (s, 9H), 1.42 (t, 2H), 3.10 (m, 2H), 4.14 (m, 2H), 4.67 (s, 1H), 5.01 (m, 1H), 5.79 (d, 1H), 6.07 (dd, 1H), 6.37 (d, 1H); 13C NMR (CDCl3) δ (ppm): 14.1, 17.0, 22.7, 26.5, 30.0, 31.6, 41.2, 66.6, 68.6, 128.2, 131.4, 156.0, 166.0. Elemental analysis: found: C, 60.5; H, 9.0; N, 5.5. C13H23NO4 requires: C, 60.9; H, 8.7; N, 5.5%.Synthesis of polymer standards
The synthesis of standards for poly(PhCEA) is described. The procedures are the same for all monomers under investigation. Two general methods were used for molecular weight control: (i) control vian-octanethiol as a chain transfer agent and (ii) control of molecular weight viainitiator concentration.(i) A solution of PhCEA (5.9641 g, 0.025 mol) in butyl acetate (25 mL) was made up. Then, solutions of AIBN (0.0049 g, 3.0 × 10−5 mol) and n-octanethiol (varying, ≤0.08 g) in butyl acetate (10 mL) were prepared and the final reaction solutions were obtained from mixing 3 mL monomer solution and 1 mL AIBN–n-octanethiol solution. (ii) AIBN (0.1640 g, 1.0 × 10−3 mol) was dissolved in ethyl acetate (10 mL). Lower concentrations of this solution were obtained by successive dilutions to give [AIBN] in the range 1 × 10−3 to 0.1 mol L−1. To 2 mL of these solutions, PhCEA (approximately 0.4705 g) was then dissolved.
The reaction solutions were placed in glass vials, sealed with rubber septa and purged with nitrogen for 5 min before being reacted at 60 °C for 90 min. Reactions were quenched by exposure to oxygen, and the solvent was left for evaporation. Subsequently, residual monomer was removed viadialysis in acetone, using SpectraPor #3 DialysisMembranes (molecular weight cut-off 3500 Da).
Absolute molecular weight determination
The triple-detection chromatographic setup used for the determination of the MHKS parameters consisted of a modular system (Polymer Standard Service, PSS, Mainz/Agilent 1200 series) incorporating an ETA2010 viscometer (WGE Dr Bures) and a MALLS light-scattering unit (PSS SLD7000/BI-MwA, Brookhaven Instruments). Sample separation is achieved via two linear columns provided by PSS (SDV-Lux-1000 Å and 105 Å, 5 µm) with THF as the eluent at 35 °C with a flow rate of 1 mL min−1. The system was calibrated using polystyrene standards (PSS). The sample concentration typically was close to 3 mg mL−1. dn/dc was determined as the average from all synthesized standards.Pulsed laser polymerization
Solutions of the monomers in butyl acetate solution (with typical monomer concentrations of 1 mol L−1) containing 5 × 10−3 mol L−1DMPA were transferred into sample vials (containing about 0.2 mL of reaction solution each) and sealed with rubber septa. Oxygen was removed by purging the samples with nitrogen for about 2 min. The sample vial was then placed into a stainless steel sample holder that was temperature controlled by a thermostat (VWR 1196D). Temperature was measured directly in the sample. The samples were allowed to equilibrate in temperature for about 5 min and were subsequently initiated by laser pulsing at constant repetition rates of up to 500 Hz. The number of laser pulses n applied is given for each sample individually in Tables 2–5. Laser initiation was achieved by a Xantos XS-500 operated at 351 nm. The laser beam, which was adjusted to an energy of close to 2 mJ per pulse hitting the sample, was redirected to illuminate the vial from the bottom. After polymerization, hydroquinone–methanol solution was added to the samples. Methanol and butyl acetate were then evaporated and the monomer–polymer mixture was directly submitted to SEC analysis. Monomer conversion was checked via1H NMR.Molecular weight determination
For the determination of molecular weight distributions (MWDs) obtained from PLP, a Varian system, comprising an auto injector, a Polymer Laboratories 5.0 µm bead-size guard column, followed by three linear PL columns (PLgel 5 µm MIXED-C) and a differential refractive index detector using THF as the eluent at 40 °C with a flow rate of 1 mL min−1 was used. The SEC system was calibrated using narrow polystyrene standards ranging from 160 to 6 × 106 g mol−1. The resulting molecular weight distributions have been recalibrated employing the Mark–Houwink–Kuhn–Sakurada parameters of the corresponding polymer and for polystyrene (K = 14.1 × 10−5 dL g−1 and a = 0.70).Results and discussion
MHKS parameter determination
Knowledge of the correct MHKS parameters of the polymer under investigation is crucial for PLP-SEC measurements. Most molecular weight analysis relies on the principle of universal calibration, which is based on the advantage of polymers with identical intrinsic viscosity eluting at the same retention volume regardless of the nature of the polymer type. Without the parameters K and a (see eqn (1)) that connect the viscosity of polymer coils in the respective solvent [η] with molecular weight, M, true molecular weights from standard SEC (that is chromatography systems equipped with concentration sensitive detection only) are thus not available and only qualitative results can be obtained without them. Universal calibration in conjunction with the MHKS relation is a relatively robust method within certain boundaries. Its applicability is restricted to polymers of sufficient length enabling them to form coils and one set of MHKS parameters are strictly only valid for defined topologies. As a consequence, universal calibration is only safe to employ from molecular weights above 5–10 × 103 Da and in cases where chains are strictly non-branched.| log[η] = log K + a × log M | (1) |
For acrylates, these constrains are hard to meet. As noted above, extensive transfer to polymer reactions lead to a significant number of chain branches on the polymer chains. Thus, the number and type of branches depend on the synthetic procedure and on the reaction conditions. MHKS parameters determined from a set of polyacrylates are only valid for polymer that was synthesized under similar reaction conditions. With regard to polymers generated viaPLP, an even more complicated situation exists because it must be assumed that polymer that was made from laser pulsing is not homogeneous in its branching structure (for a more thorough discussion on the specific consequences of PLP on branching, see ref. 9 and 24). The laser action interrupts the quasi-equilibration between mid-chain and secondary radicals and thus the average topology of the chains depends not only on the reaction temperature but also on the laser pulse repetition rate.
Nevertheless, while branches are generated throughout the entire growth of a single chain, a few assumptions can be made: upon the laser action, the system is flooded with small, initiator-derived radicals that terminate all radicals irrespective of their nature. Mid-chain radicals present at time t0 (see eqn (2)) are quenched and no chain branch is obtained. Thus, PLP promotes the formation of linear chains specifically at the characteristic chain lengths that will later be analyzed. Secondly, the time span between two consecutive laser pulses is short and most of the chains are terminated before a larger number of branches can be formed. For example, at 60 °C, a backbiting rate coefficient kbb of close to 100 s−1 is expected.31 Thus, when laser pulses are fired at 500 Hz, only about 20% of all chains statistically had the chance to undergo one transfer step potentially forming a chain branch. (The validity of this assumption is directly evident from the experiment: the formation of chain branches by mid-chain radical propagation is slow, hence backbiting destroys the time–chain length relation. Thus, if the characteristic chain length distribution of a PLP sample is obtained, only a small number of transfer events can have taken place.) In summary, the polymer that is formed in the duration between two laser pulses is most likely of linear topology and only chains that survive a multiple of laser pulses will be affected by branching with higher probability.
Under the consideration of the above points we have chosen to synthesize polymer standards by free radical polymerization under mild reaction conditions, i.e. at as low as possible reaction temperatures. To synthesize polymers with controllable average molecular weight, standards were (partially) polymerized under addition of small amounts of thiol, which should also prevent formation of branching points to a significant degree.32,33 While the MHKS values that are determined on such samples should reflect PLP samples on average, such an approach is still a compromise, also with respect to the polymerization temperature. It should be noted that it is advisable to employ triple-detection SEC for all PLP samples generated from acrylic monomers. The fact that we use universal calibration despite all concerns has two reasons: (i) universal calibration allows for a simple and fast determination of molecular weights. (ii) The monomers under investigation are all solid at ambient temperature, hence a separation of polymer from monomer viadialysis in an organic solvent is tedious and in consequence determination of molecular weight vialight scattering and viscosimetry rather difficult for the large number of generated PLP samples. Additionally, while it is apparent that the occurrence of branching has a damaging effect for the application of universal calibration, it is unclear how large the deviation is, hence the error in molecular weight determination may be tolerable. In addition, an indication exists that—at least in some cases—the temperature at which the PLP experiment was carried out has only a minor influence on the MHKS parameters.34 Further, it should not be forgotten that even under conditions of perfect calibration, SEC is associated with an error of up to 20%.
Fig. 1 displays plots of the average intrinsic viscosity vs. average molecular weight for polymer standards of all four monomers given in Scheme 1. Satisfactory linear relations following eqn (1) are observed in all cases. The resulting K and a values are collated in Table 1 together with the respective dn/dc values for the polymers. For both phenyl derivatives relatively similar results are obtained. The additional methyl group has apparently no large influence on the viscosity of the coils in the solventTHF. A qualitatively similar trend is observed when comparing both hexyl compounds, where also relatively similar values for K and a are observed. Between both phenyl and hexyl isocyanate based monomers, however, a profound difference is seen in that the hexyl group leads to a significant decrease in a and thereby increase of K. All plots show satisfactory linear dependencies, thus it is reasonable to assume that the quality of the determined MHKS values is relatively high, especially as they are based on several different polymer samples. The fact that the fits are relatively good is also seen by the small error margins that are derived from the regressions. Only HCEA shows a somewhat larger error for K and a due to the somewhat smaller mass range that was covered from the synthesized standards. It should hereby be noted that synthesis of polymer standards with higher molecular weight was attempted, however, even under polymerization conditions without control agent and with low amounts of initiator, no poly(HCEA) with higher molar mass could be obtained.
| PhCEA | PhCPA | HCEA | HCPA | |
|---|---|---|---|---|
| 105K/dL g−1 | 8.3 ± 1.1 | 10.3 ± 2.0 | 36.0 ± 16 | 28.0 ± 6.9 |
| a | 0.677 ± 0.012 | 0.657 ± 0.015 | 0.552 ± 0.037 | 0.587 ± 0.019 |
| dn/dc/mL g−1 | 0.13 | 0.12 | 0.073 | 0.071 |
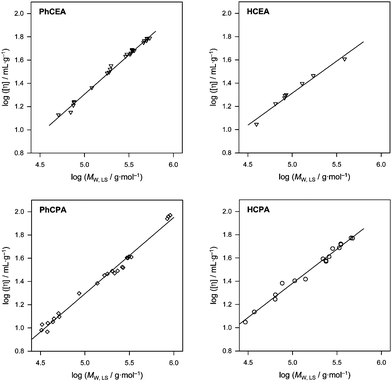 | ||
| Fig. 1 Intrinsic viscosity of polymer samples as a function of average molecular weight for the determination of MHKS values. | ||
Pulsed laser polymerization
The basic principle of the pulsed laser polymerization technique is based on the linear growth of radicals after the virtually instantaneous generation of a population of radicals by the action of a laser pulse. The majority of growing radicals are terminated by the following laser pulse and thus predominantly polymer of time–specific chain length is generated. By analysis of the resulting molecular weight distributions, the propagation rate coefficient kp can be directly derived from the (local) maximum of the distribution. Because a small fraction of radicals survives the quenching by a consecutive laser pulse, however, multimodal distributions are often obtained where the individual maxima correspond to a multitude of dark times and thus the propagation rate coefficient can be calculated from different points of the distribution. Thus, PLP-SEC not only allows for the determination of kp, it also provides an inherent measure for the quality of the experiment and hence reliability of the result.| Li = kp × cM × i × t0, where i = 1, 2, 3,… | (2) |
In practice, the characteristic points of the distributions are best determined by identifying the inflection points Li from the first derivative of the MWD (it should be noted that Li refers to the chain length rather than mass). An example is shown in Fig. 2 which depicts the MWD obtained from PLP of HCPA at 500 Hz laser repetition rate and a temperature of 14.1 °C. Up to four inflection points can clearly be distinguished from the maxima of the derivative curve. Inspection of the derivative allows for the accurate determination of kpviaeqn (2) from all four characteristic points where t0 refers to the dark time between two consecutive laser pulses, i is the number of dark time periods that the radical survived and cM is the initial monomer concentration.
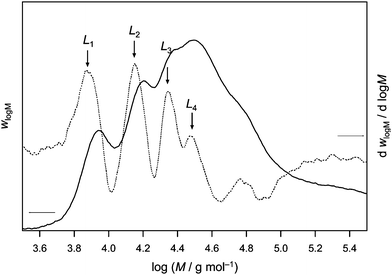 | ||
| Fig. 2 Molecular weight distribution as determined viaSEC and its first derivative of a polyHCPA sample made by PLP at 14.1 °C. | ||
2-(Phenylcarbamoyloxy)ethyl acrylate, PhCEA
Pulsed laser experiments were performed on PhCEA between 5 and 70 °C on a 1 M solution of the monomer in butyl acetate. Due to the crystallinity of the monomers, two problems occur. Firstly, PLP needs to be carried out at low monomer conversion to ensure that the position of the inflection points is not affected by the progress of the polymerization (and thus a decrease in cM). Gravimetry cannot be directly employed to determine conversions and spectroscopic online methods such as FT-NIR were not available, thus some samples have been subjected to 1H NMR analysis. No sample showed significant contribution of polymeric peaks and basically spectra of the monomer were obtained. Thus, it was concluded that all samples taken at a similar number of laser pulses were polymerized to conversions lower than 5%.The other problem is that PLP is preferentially carried out on bulk solutions. It is known that the polarity of the reaction medium can have an influence on the propagation rate and thus solvents are usually avoided.12PLP-SEC on the monomer in the melt could not be carried out due to the relatively high melting points (close to 70 °C depending on the monomer). Thus, polymerizations were carried out in solution of butyl acetate, a solvent that should be comparable in polarity to the monomer and hence only small influences from the solvent are expected. Nevertheless, it must be taken into account that a small influence may exist.
All experimental results for PhCEA are collated in Table 2 and plotted in Fig. 3. The table summarizes the results from L1 and L2 (with Li = Mi/Mmonomer). Up to 4 inflection points were observed in some cases; however, the number of observable inflection points decreases with increasing temperature. Especially at lower temperatures, kp can be consistently calculated from any of the inflection points. However, the data presented herein are all based on the first point of inflection and Fig. 3 (as in the Arrhenius plots for all following monomers) only depicts kp,1. As a consistency check, only data were considered where kp,1 did not show any higher deviation from kp,2 than 10% at maximum.
| T/°C | n | log M1/g mol−1 | log M2/g mol−1 | log M3/g mol−1 | k p,1 /L mol−1 s−1 | k p,2 /L mol−1 s−1 | k p,1 /kp,2 |
|---|---|---|---|---|---|---|---|
| 4.8 | 200 | 4.065 | 4.346 | 4.534 | 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 690 690 |
23570 |
1.05 |
| 5 | 350 | 4.065 | 4.346 | 4.534 | 24690 |
23570 |
1.05 |
| 9.5 | 200 | 4.116 | 4.426 | 4.623 | 27770 |
28370 |
0.98 |
| 20.0 | 250 | 4.206 | 4.522 | 4.734 | 34190 |
35350 |
0.97 |
| 20.0 | 100 | 4.195 | 4.516 | 4.729 | 33310 |
34900 |
0.95 |
| 20.8 | 50 | 4.201 | 4.528 | 4.734 | 33750 |
35810 |
0.94 |
| 29.7 | 50 | 4.280 | 4.606 | 4.823 | 40480 |
42900 |
0.94 |
| 30.0 | 150 | 4.280 | 4.606 | 4.817 | 40480 |
42900 |
0.94 |
| 31 | 150 | 4.248 | 4.556 | 4.758 | 37590 |
38260 |
0.98 |
| 40.0 | 80 | 4.359 | 4.684 | 4.900 | 48550 |
51350 |
0.95 |
| 40.5 | 60 | 4.359 | 4.679 | 4.894 | 48550 |
50700 |
0.96 |
| 50.2 | 80 | 4.443 | 4.745 | 4.965 | 58930 |
59050 |
1.00 |
| 50.3 | 100 | 4.449 | 4.756 | — | 59690 |
60580 |
0.99 |
| 60.3 | 50 | 4.516 | 4.801 | — | 69710 |
67070 |
1.04 |
| 61.2 | 100 | 4.522 | 4.806 | — | 70630 |
67930 |
1.04 |
| 70.4 | 50 | 4.578 | 4.861 | — | 80360 |
77110 |
1.04 |
| 70.5 | 100 | 4.567 | 4.839 | — | 78320 |
73300 |
1.07 |
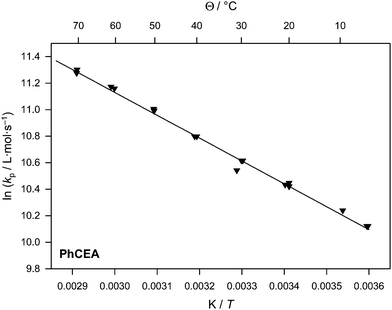 | ||
| Fig. 3 Arrhenius plot of the propagation rate coefficient for PhCEA. | ||
The high linearity of the data in Fig. 3 demonstrates the overall high quality of the obtained kp data. Only little scatter can be observed and the Arrhenius fit yields an activation energy EA for kp of 14.3 kJ mol−1 and a frequency factor of A = 1.2 × 107 L mol−1 s−1. If compared to n-butyl acrylate, a linear alkyl analogue of PhCEA, a comparatively high-frequency factor is observed (A(BA) = 2.1 × 107 L mol−1 s−1 with EA(BA) = 17.8 kJ mol−1) while the activation energy is significantly reduced.22 Thus, larger kp values are obtained for PhCEA in the temperature range under investigation compared to the n-butyl ester. For example at 60 °C, kp of the carbamate is almost a factor of two larger. Given that the phenyl carbamate moiety is relatively bulky and not very flexible, this result is remarkable. An increase of the propagation rate coefficient is generally observed within the acrylate and other monomer families, however, only for linear alkyl esters. This rate increase is today explained by a shielding of the polar estergroup, which decreases the friction of internal rotation in the transition state of the reaction. From this friction the frequency factor of the reaction is influenced. However, in the present case, the increase of the propagation rate is not based on a larger frequency factor, but on a reduced activation energy, especially as the low flexibility of the carbamate functionality should prevent effective shielding of the dipolar interactions between two ester moieties. In contrast, it must be hypothesized that the decrease in the activation barrier has electronic effects—or is influenced by H-bonding which could also allow for a smaller energy gap between the transition and ground state. It is noteworthy to add that a very similar decrease of the activation energy in conjunction with an overall increased kp was recently observed for the monomer ethoxy ethyl acrylate (EEA).24EEA does not contain a carbamate moiety, however, it has in common with the carbamate acrylates that its structure is based on hydroxylethyl acrylate and hence carries an ethylene glycol bridge. Interestingly, Kilambi et al. have shown that such substituents lower the LUMO energy of an acrylate and in consequence increase their nucleophilicity, which in return lowers the activation energy of the propagation reaction.20 While EEA displayed a similar activation energy (13.8 kJ mol−1) compared to PhCEA, the frequency factor was smaller by a factor of approximately two, so the ability to form H-bonds or solvent interactions may have an additional rate-increasing influence.
2-(Phenylcarbamoyloxy)isopropyl acrylate, PhCPA
When the ethyl bridge of the monomer is methylated, a reduction in the overall propagation rate is observed. Table 3 and Fig. 4 depict the outcome from PLP-SEC experiments of PhCPA in the temperature range from 5 to 80 °C. Again, the quality of the data is relatively high. Excellent agreement between kp,1 and kp,2 is observed, even at higher temperatures where the distributions are more severely blurred due to the increased transfer activity. Again, a reduced activation energy is obtained compared to alkyl acrylates.4,22,24 For PhCPA, EA = 14.2 kJ mol−1 and A = 4.9 × 106 L mol−1 s−1 are obtained. Thus, at any temperature kp is decreased by a constant factor of 2.4 by introducing the additional methyl group. Despite such a rate decrease, kp(PhCPA) is still in the range of alkyl acrylates as can be seen from the relatively similar value for 60 °C (for a comparison of the activation parameters and kp at 60 °C for the monomers under investigation in the current study and for n-butyl acrylate see Table 6). Because the monomer actually consists of a mixture of isomers (see Scheme 2), it is difficult to judge what exactly causes this decrease. It may, however, be assumed that the additional methyl group results in more rigid conformations and generally less flexibility of the polymer chains. In any case, the decrease in the overall polymerization rate for the methylated carbamate acrylates as observed by Bowman and co-workers15 may thus be understood on the basis of the propagation rate (and potentially influences on the termination rate, which most likely is also reduced by the increased stiffness of the chains).| T/°C | n | log M1/g mol−1 | log M2/g mol−1 | log M3/g mol−1 | k p,1 /L mol−1 s−1 | k p,2 /L mol−1 s−1 | k p,1 /kp,2 |
|---|---|---|---|---|---|---|---|
| 5.5 | 250 | 4.080 | 4.367 | 4.572 | 11000 |
10650 |
1.03 |
| 5.5 | 110 | 4.072 | 4.359 | 4.572 | 10800 |
10460 |
1.03 |
| 9.5 | 250 | 4.106 | 4.399 | 4.601 | 11690 |
11460 |
1.02 |
| 9.5 | 150 | 4.103 | 4.395 | 4.597 | 11590 |
11360 |
1.02 |
| 9.9 | 100 | 4.099 | 4.395 | 4.593 | 11490 |
11360 |
1.01 |
| 19.0 | 130 | 4.195 | 4.503 | 4.696 | 14330 |
14570 |
0.98 |
| 19.0 | 90 | 4.187 | 4.499 | 4.692 | 14080 |
14440 |
0.98 |
| 20.1 | 200 | 4.199 | 4.507 | 4.700 | 14460 |
14710 |
0.98 |
| 29.9 | 200 | 4.288 | 4.601 | 4.793 | 17770 |
18250 |
0.97 |
| 30.3 | 80 | 4.288 | 4.597 | 4.797 | 17770 |
18080 |
0.98 |
| 31.0 | 130 | 4.292 | 4.601 | 4.797 | 17930 |
18250 |
0.98 |
| 40.5 | 90 | 4.367 | 4.679 | 4.887 | 21300 |
21870 |
0.97 |
| 40.9 | 100 | 4.379 | 4.688 | 4.900 | 21900 |
22290 |
0.98 |
| 41.4 | 150 | 4.387 | 4.688 | 4.596 | 22300 |
22300 |
1.00 |
| 50.2 | 80 | 4.463 | 4.755 | 4.978 | 26560 |
26020 |
1.02 |
| 50.3 | 100 | 4.471 | 4.751 | 4.982 | 27060 |
25760 |
1.05 |
| 60.2 | 40 | 4.521 | 4.792 | — | 26910 |
25130 |
1.07 |
| 70.5 | 30 | 4.616 | 4.961 | — | 33460 |
37060 |
0.90 |
| 70.8 | 70 | 4.595 | 4.935 | — | 31900 |
34890 |
0.91 |
| 80.2 | 60 | 4.708 | 5.005 | — | 41340 |
41020 |
1.01 |
| 81.1 | 40 | 4.687 | 4.992 | — | 39390 |
39790 |
0.99 |
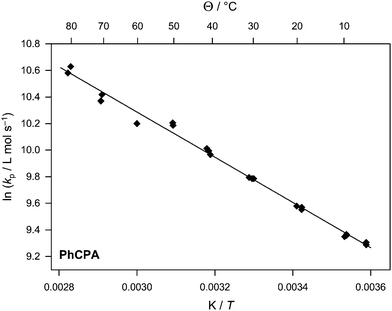 | ||
| Fig. 4 Arrhenius plot of the propagation rate coefficient for PhCPA. | ||
2-(Hexylcarbamoyloxy)ethyl acrylate, HCEA
For HCEA qualitatively similar results are obtained as for the other monomers and the resulting propagation rate coefficients are collated in Table 4 and in Fig. 5. The temperature range that is covered and where a clear PLP pattern was still observable is between 5 and 70 °C. Fitting the data yields an activation energy of 13.3 kJ mol−1 and a frequency factor of A = 6.6 × 106 L mol−1 s−1. Thus, kp is relatively high and at 60 °C, kp = 54080 L mol−1 s−1 is obtained, which is somewhat smaller than kp of PhCEA. The activation energy is slightly smaller than for the other monomers, however, a remark on the accuracy of the result should be given here. The error estimates in Table 6 refer to the statistical error from fitting the data, thus assuming the determination of molecular weights via conventional SEC is practically error free. Of course, as is evident from the MHKS plots given in Fig. 1, universal calibration is beset with an uncertainty, let alone that SEC even with direct calibration is not error free. Thus, errors in the MHKS parameters must in principle be considered when stating an error for the Arrhenius parameters of kp. Such estimation is not completely straightforward as the error might be different for various molecular weight ranges. Also, the error may depend on whether the standards that were used for MHKS determination were suitable representatives for the later PLP samples (see the discussion above). Thus, the true error can only be approximately estimated and an uncertainty of about 1 kJ mol−1 for the activation energy should be assumed for all four monomers under investigation. Therefore, EA(HCEA) is within error limits identical to the activation energies of the other monomers. The fact that kp is slightly smaller than kp of PhCEA shows that the hexyl substituent has a significant effect on the propagation rate. The exact reason for this rate reduction cannot be elucidated from the present data, but a potential explanation is that the hexyl group has a larger screening radius and hence the approach of a monomer molecule to the chain end of a growing chain may be slightly more hindered when compared to the phenyl analogue.
| T/°C | n | log M1/g mol−1 | log M2/g mol−1 | log M3/g mol−1 | k p,1 /L mol−1 s−1 | k p,2 /L mol−1 s−1 | k p,1 /kp,2 |
|---|---|---|---|---|---|---|---|
| 5 | 200 | 4.105 | 4.435 | 4.646 | 20990 |
22390 |
0.94 |
| 5.3 | 250 | 4.112 | 4.447 | 4.652 | 21290 |
23030 |
0.92 |
| 5.4 | 130 | 4.105 | 4.435 | 4.646 | 20990 |
22390 |
0.94 |
| 9.2 | 150 | 4.136 | 4.465 | 4.676 | 22520 |
24020 |
0.94 |
| 9.3 | 100 | 4.130 | 4.465 | 4.676 | 22210 |
24020 |
0.92 |
| 9.6 | 200 | 4.130 | 4.465 | 4.670 | 22210 |
24020 |
0.92 |
| 19.9 | 50 | 4.203 | 4.538 | 4.742 | 26280 |
28390 |
0.93 |
| 20 | 300 | 4.203 | 4.538 | 4.742 | 26280 |
28390 |
0.93 |
| 20.3 | 200 | 4.191 | 4.526 | 4.736 | 25550 |
27610 |
0.93 |
| 21 | 100 | 4.203 | 4.538 | 4.748 | 26280 |
28390 |
0.93 |
| 30 | 80 | 4.282 | 4.604 | 4.808 | 31650 |
33200 |
0.95 |
| 32 | 180 | 4.307 | 4.622 | 4.826 | 33480 |
34610 |
0.97 |
| 32.4 | 350 | 4.301 | 4.622 | 4.820 | 33010 |
34610 |
0.95 |
| 32.4 | 250 | 4.288 | 4.610 | 4.808 | 32100 |
33660 |
0.95 |
| 41.3 | 250 | 4.398 | 4.700 | 4.909 | 41310 |
41430 |
1.00 |
| 41.4 | 200 | 4.386 | 4.688 | 4.897 | 40170 |
40300 |
1.00 |
| 41.5 | 90 | 4.386 | 4.694 | 4.903 | 40170 |
40860 |
0.98 |
| 41.7 | 50 | 4.380 | 4.688 | 4.915 | 39600 |
40300 |
0.98 |
| 41.7 | 110 | 4.386 | 4.694 | 4.311 | 40170 |
40860 |
0.98 |
| 50.3 | 100 | 4.447 | 4.748 | 4.979 | 46060 |
46120 |
1.00 |
| 50.3 | 50 | 4.441 | 4.748 | 4.979 | 45420 |
46120 |
0.98 |
| 50.7 | 130 | 4.447 | 4.742 | 4.968 | 46060 |
45490 |
1.01 |
| 60.3 | 100 | 4.507 | 4.790 | — | 52960 |
50780 |
1.04 |
| 60.3 | 50 | 4.507 | 4.802 | — | 52960 |
52190 |
1.01 |
| 60.4 | 80 | 4.507 | 4.796 | — | 52960 |
51480 |
1.03 |
| 62.2 | 200 | 4.513 | 4.790 | — | 53710 |
50780 |
1.06 |
| 70.3 | 60 | 4.562 | 4.844 | — | 60040 |
57440 |
1.05 |
| 70.5 | 30 | 4.568 | 4.844 | — | 60880 |
57440 |
1.06 |
| T/°C | n | log M1/g mol−1 | log M2/g mol−1 | log M3/g mol−1 | k p,1 /L mol−1 s−1 | k p,2 /L mol−1 s−1 | k p,1 /kp,2 |
|---|---|---|---|---|---|---|---|
| 2.8 | 250 | 3.867 | 4.151 | 4.354 | 14250 |
13710 |
1.04 |
| 2.8 | 200 | 3.879 | 4.159 | 4.375 | 14650 |
13970 |
1.05 |
| 13.7 | 100 | 3.978 | 4.270 | 4.477 | 18400 |
18050 |
1.02 |
| 14.1 | 200 | 3.962 | 4.258 | 4.464 | 17740 |
17540 |
1.01 |
| 27.6 | 100 | 4.090 | 4.404 | 4.598 | 23840 |
24570 |
0.97 |
| 27.7 | 200 | 4.090 | 4.421 | 4.611 | 23840 |
25550 |
0.93 |
| 40.6 | 100 | 4.192 | 4.498 | 4.708 | 30130 |
30500 |
0.99 |
| 50.2 | 120 | 4.258 | 4.558 | 4.766 | 35070 |
34990 |
1.00 |
| 51.2 | 150 | 4.266 | 4.559 | 4.811 | 35750 |
35040 |
1.02 |
| 60.2 | 80 | 4.350 | 4.646 | 4.874 | 43320 |
42850 |
1.01 |
| 61.5 | 120 | 4.329 | 4.633 | — | 41280 |
41570 |
0.99 |
| 70.6 | 80 | 4.447 | 4.681 | — | 54190 |
46480 |
1.17 |
| 72.4 | 120 | 4.434 | 4.664 | — | 52620 |
44630 |
1.18 |
| PhCEA | PhCPA | HCEA | HCPA | BA | |
|---|---|---|---|---|---|
| ln A/L mol−1 s−1 | 16.3 ± 0.1 | 15.4 ± 0.1 | 15.7 ± 0.1 | 15.7 ± 0.1 | 16.9 |
| E A/kJ mol−1 | 14.3 ± 0.3 | 14.2 ± 0.3 | 13.3 ± 0.2 | 14.1 ± 0.2 | 17.8 |
| k p (60 °C) | 68680 |
28950 |
54080 |
40510 |
34470 |
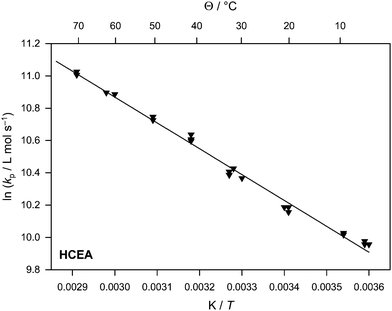 | ||
| Fig. 5 Arrhenius plot of the propagation rate coefficient for HCEA. | ||
2-(Hexylcarbamoyloxy)isopropyl acrylate, HCPA
Again, with HCPA, PLP allowed for the generation of well structured molecular weight distributions with multiple inflection points. PLP patterns were observed up to 70 °C. However, when the results from the first two inflection points of the samples taken at 70 °C are compared, a difference of more than 15% between both values is observed. Thus, as only data that showed 5–10% maximum deviation are considered in the present study—in line with the recommendations of the IUPAC for reliable PLP-SEC experiments—only the data taken for temperatures from 5 to 60 °C were fitted to obtain the activation parameters of kp. Nevertheless, even if the data taken for 70 °C were included in the fit, no significantly different result is obtained. From the fit as shown in Fig. 6 (based on the data given in Table 5) an activation energy of 14.1 kJ mol−1 and a frequency factor of 6.6 × 106 L mol−1 s−1 are deduced for kp(HCPA). Thus, kp is at any temperature by approximately 30% smaller than kp of HCEA. This observation is in good agreement with the trend seen between the two phenyl derivatives, where also a significantly reduced kp was observed when going from the ethyl to the isopropyl bridge. However, the decrease is not as pronounced as for the phenylcarbamates and in fact, kp(T) of HCPA is above kp(T) of PhCPA. Thus, even though the hexyl group has only a small influence on kp when PhCEA and HCEA are compared, it can apparently significantly counteract the influence of the additional methyl group. Such a behaviour can again be understood on the basis of the above hypothesis that the isopropyl group leads to a less flexible ester side chain whereby the hexyl group—while introducing more steric hindrance—leads to a higher flexibility. However, further proof is required for this hypothesis.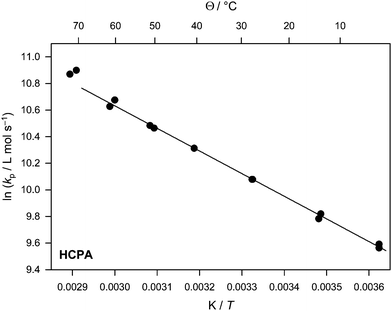 | ||
| Fig. 6 Arrhenius plot of the propagation rate coefficient for HCPA. | ||
Conclusions
All four monomers under investigation are comparatively rapidly propagating monomers, despite the relatively high bulkiness of the ester side chains. Thus, the generally high overall polymerization rates that are observed with these types of monomers can at least partially be attributed to a significant increase in the propagation rate when compared to other acrylate monomers. The fast propagating nature of the monomers may be understood on the basis of the increased nucleophilicity that is induced by the carbamate functionality present in all monomers. Rate-increasing effects from solvent polarity and/or from H-bonding can, however, not be excluded and might also contribute to the observed high rates. Generally, exchanging the phenyl group for a hexyl group leads to a decrease in the propagation rate. Interestingly, a similar observation was made before when the propagation rate of cyclic vs. linear methacrylate esters was compared, thus cyclic groups may have a general rate enhancing effect.4 A rate decrease is also observed when the ethyl bridge in the monomer is replaced by an isopropyl group. Introducing the isopropyl group, however, has a larger effect than varying the phenyl/hexyl group. Overall, PhCEA is found to be a rapidly growing monomer when compared to linear alkyl acrylates and further studies focusing on the solvent influence on the propagation rate appear to be expedient. All four monomers show similar activation energies with 14 ± 1 kJ mol−1, which is lower than what is usually found for alkyl acrylates by about 4 kJ mol−1. In consequence, the difference in absolute kp and thus the overall polymerization rate between the herein studied monomers and alkyl acrylates is significantly larger at reduced temperature, e.g. ambient temperature. Such a decreased activation barrier is in agreement with a previous study,24 where the monomer ethoxy ethyl acrylate, whose structure is also based on hydroxyethyl acrylate, also featured a similar low activation energy.Acknowledgements
C.B.-K. acknowledges financial support from the Karlsruhe Institute of Technology (KIT) within the Excellence Initiative for leading German Universities supporting the current project. C.B.-K. is grateful for additional support for the current project from Bayer MaterialScience AG. T.J. is grateful for additional support from the Fonds der Chemischen Industrie.References
- A. P. Aleksandrov, V. N. Genkin, M. S. Kitay, I. M. Smirnova and V. V. Sokolov, Sov. J. Quantum Electron., 1977, 5, 976–981.
- O. F. Olaj, I. Bitai and F. Hinkelmann, Makromol. Chem., 1987, 188, 1689–1702 CrossRef CAS.
- O. F. Olaj and I. Schnöll-Bitai, Eur. Polym. J., 1989, 25, 635–641 CrossRef CAS.
- S. Beuermann and M. Buback, Prog. Polym. Sci., 2002, 27, 191–254 CrossRef CAS.
- M. Buback, R. G. Gilbert, R. A. Hutchinson, B. Klumpermann, F. D. Kuchta, K. F. O'Driscoll, G. T. Russell and J. Schweer, Macromol. Chem. Phys., 1995, 196, 3267–3280 CrossRef CAS.
- S. Beuermann, M. Buback, T. P. Davis, R. G. Gilbert, R. A. Hutchinson, O. F. Olaj, G. T. Russell, J. Schweer and A. M. van Herk, Macromol. Chem. Phys., 1997, 198, 1545–1560 CrossRef CAS.
- S. Beuermann, M. Buback, T. P. Davis, R. G. Gilbert, R. A. Hutchinson, A. Kajiwara, B. Klumpermann and G. T. Russell, Macromol. Chem. Phys., 2000, 201, 1355–1364 CrossRef CAS.
- S. Beuermann, M. Buback, T. P. Davis, R. G. Gilbert, R. A. Hutchinson, A. Kajiwara, M. Kamachi, I. Lacik and G. T. Russell, Macromol. Chem. Phys., 2003, 204, 1338–1350 CrossRef CAS.
- J. M. Asua, S. Beuermann, M. Buback, P. Castignolles, B. Charleux, R. G. Gilbert, R. A. Hutchinson, J. R. Leiza, A. N. Nikitin, J. P. Vairon and A. M. van Herk, Macromol. Chem. Phys., 2004, 205, 2151–2160 CrossRef CAS.
- S. Beuermann, M. Buback, P. Hesse, F. D. Kuchta, I. Lacik and A. M. van Herk, Pure Appl. Chem., 2007, 79, 1463–1469 CrossRef CAS.
- (a) T. Gruendling, M. Guilhaus and C. Barner-Kowollik, Macromolecules, 2009, 42, 6366–6374 CrossRef CAS; (b) T. Gruendling, M. Guilhaus and C. Barner-Kowollik, Anal. Chem., 2008, 80, 6915–6927 CrossRef CAS; (c) T. Gruendling, D. Voll, M. Guilhaus and C. Barner-Kowollik, Macromol. Chem. Phys., 2010, 211, 80–90 CrossRef CAS.
- S. Beuermann, Macromol. Rapid Commun., 2009, 30, 1066–1088 CrossRef CAS.
- S. Beuermann, M. Buback, P. Hesse and I. Lacik, Macromolecules, 2006, 39, 184–193 CrossRef CAS.
- S. Beuermann and D. Nelke, Macromol. Chem. Phys., 2003, 204, 460–470 CrossRef CAS.
- E. R. Beckel, J. W. Stansbury and C. N. Bowman, Macromolecules, 2005, 38, 3093–3098 CrossRef CAS.
- H. Kilambi, E. R. Beckel, K. A. Berchtold, J. W. Stansbury and C. N. Bowman, Polymer, 2005, 46, 4735–4742.
- H. Kilambi, D. Konopka, J. W. Stansbury and C. N. Bowman, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1287–1295 CrossRef CAS.
- H. Kilambi, J. W. Stansbury and C. N. Bowman, Macromolecules, 2007, 40, 47–54 CrossRef CAS.
- N. B. Cramer, C. P. O'Brien and C. N. Bowman, Polymer, 2008, 49, 4756–4761 CrossRef CAS.
- H. Kilambi, S. K. Reddy, L. Schneidewind, J. W. Stansbury and C. N. Bowman, Macromolecules, 2009, 42, 4859–4870.
- W. Fischer, J. Weikard, E. Luhmann and T. Fäcke, Eur. Coat. J., 2003, 11, 28–30 Search PubMed.
- C. Barner-Kowollik, F. Günzler and T. Junkers, Macromolecules, 2008, 41, 8971–8973 CrossRef CAS.
- T. Junkers, D. Voll and C. Barner-Kowollik, e-polymers, 2009, 076 Search PubMed.
- B. Dervaux, T. Junkers, M. Schneider-Baumann, F. E. Du Prez and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6641–6654 CrossRef CAS.
- T. Junkers and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7585–7605 CrossRef CAS.
- L. Couvreur, G. Piteau, P. Castignolles, M. Tonge, B. Coutin, B. Charleux and J. P. Vairon, Macromol. Symp., 2001, 174, 197–207 CrossRef CAS.
- A. N. Nikitin and R. A. Hutchinson, Macromolecules, 2005, 38, 1581–1590 CrossRef CAS.
- (a) P. J. Flory, J. Am. Chem. Soc., 1937, 59, 241–253 CrossRef CAS; (b) M. J. Roedel, J. Am. Chem. Soc., 1953, 75, 6110–6112 CrossRef CAS.
- P. Castignolles, R. Graf, M. Parkinson, M. Wilhelm and M. Gaborieau, Polymer, 2009, 50, 2373–2383 CrossRef CAS.
- P. Castignolles, Macromol. Rapid Commun., 2009, 30, 1995–2001 CrossRef CAS.
- A. N. Nikitin, R. A. Hutchinson, M. Buback and P. Hesse, Macromolecules, 2007, 40, 8631–8641 CrossRef CAS.
- T. Junkers, S. P. S Koo, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Macromolecules, 2007, 40, 8906–8912 CrossRef CAS.
- S. P. S. Koo, T. Junkers and C. Barner-Kowollik, Macromolecules, 2009, 42, 62–69 CrossRef CAS.
- Detailed investigations on the quality of SEC separation of PLP samples made under various reaction conditions are currently in progress in our laboratories. At least for selected acrylates, samples made from a broad temperature range seem to obey almost the same MHKS relation, thus the effect from temperature might be negligible. Data relating to this topic will be published in a forthcoming study.
| This journal is © The Royal Society of Chemistry 2010 |