Antimicrobial polysiloxane polymers and coatings containing pendant levofloxacin
Alex
Kugel
a,
Bret
Chisholm
*ab,
Scott
Ebert
b,
Michael
Jepperson
b,
Laura
Jarabek
b and
Shane
Stafslien
b
aDepartment of Coatings and Polymeric Materials, North Dakota State University, 1735 NDSU Research Park Drive, Fargo, ND 58103, USA. E-mail: Bret.chisholm@ndsu.edu; Fax: +1 701 231 5325; Tel: +1 701 231 5328
bCenter for Nanoscale Science and Engineering, North Dakota State University, 1805 NDSU Research Park Drive, Fargo, ND 58105, USA
First published on 28th January 2010
Abstract
A broad-spectrumantimicrobial drug, levofloxacin, was successfully incorporated into a siloxane coating by covalent attachment. First, an epoxy-functional poly(dimethylsiloxane) (Ep-PDMS) was synthesized by platinum-catalyzed hydrosilylation using poly(methylhydro-co-dimethyl)siloxane and allyl glycidyl ether. Next, levofloxacin was reacted with Ep-PDMS using a stoichiometric excess of epoxy groups relative to levofloxacin to produce a siloxanecopolymer containing both pendant levofloxacin and epoxy moieties (levo-Ep-PDMS). Since attachment (i.e. tethering) of levofloxacin occurred via ring-opening of epoxy groups by the carboxylic acidgroup of levofloxacin, the tether produced was an ester-functional tether. Crosslinked surface coatings were produced by solution blending the polymer with diethylenetriamine as a crosslinker. Compared to a control coating produced by simply blending levofloxacin into a polysiloxane, the coating containing tethered levofloxacin moieties displayed a uniform distribution of levofloxacin, higher initial kill, and sustained antimicrobial surface activity.
Introduction
In today's society, advanced medical treatment involves an increasing number of procedures in which foreign materials are placed inside or in contact with the human body. For example, from 1996 to 2001, the number of hip and knee replacement surgeries increased by 14%.1 Other devices, such as venous and urethral catheters, are used daily. Whether temporary or permanent, implantation of these foreign objects into the body can facilitate transmission of microbial pathogens and cause infection in patients receiving medical treatment. The annual infection rate of implant-associated infections in the United States alone is approaching 1 million per year.2Multiple strategies have been investigated in an attempt to inhibit implant-associated infections. The approaches can be divided coarsely into systemic and local methods. Local methods of treatment provide the advantages of higher drug concentrations at or near the site of implantation3 and improved drug selection that allows use of some drugs that are ineffective or suffer toxicity when used systemically.4
Local antimicrobial prophylaxis can be generally divided into several subgroups: skin antisepsis, antimicrobial irrigation, antimicrobial carriers, dipping implants in antimicrobial solutions prior to placement in the body, and the antimicrobial coating of implants.3 The difference between these methods is the amount of time that the area remains antimicrobial. For long term solutions, antimicrobial carriers and antimicrobial coatings remain effective on the order of weeks to months while the other methods remain effective less than one day.3
Release of active materials from polymeric systems has been extensively studied.5–10 Several interrelated mechanisms drive the release process including polymer erosion, polymer swelling, and diffusion. These mechanisms are dependent on a variety of material properties such as porosity, glass transition temperature (Tg), crystallinity, molecular weight, hydrophilic/hydrophobic balance, hydrolytic stability, and crosslink density. The release rates of antimicrobial coatings and antimicrobial-doped polymers typically tend to follow first-order kinetics with a very strong release rate initially followed by an exponential decrease with time.11,12 Several experimental studies involving different antimicrobials and a broad range of polymers have been explored yielding well-known commercial products such as the MR-catheter, a catheter coated on the inner and outer surfaces with minocycline and rifampine marketed as Cook Spectrum™ catheter (Cook Critical Care, USA), and polymethylmethacrylate–gentamicin bone cement and implantable polymeric beads marketed under the Septopal® name.11–13 A variety of other material/antimicrobial combinations have been developed to effectively inhibit implant-associated infection.14 These current technologies all include the diffusion-controlled release of an active ingredient from the system resulting in a relatively short service lifetime. To extend service lifetime, covalent attachment (i.e. “tethering”) of the active ingredient to a component of the coating system using a hydrolytically labile tether is often employed. Since some preferred drugs and polymer carriers may be incompatible, tethering can also offer the added advantage of forcing compatibility and uniform dispersion of the drug throughout the polymer matrix.
One class of antimicrobial compounds that has traditionally been used systemically to combat infections is the quinolones. These materials are a class of compounds originating from a by-product of anti-malarial research in 1962 known as nalidixic acid.15 Through an incremental process over the last 40 years, nalidixic acid derivatives have grown to be an important class of antimicrobial chemicals including the well-known antimicrobial drugs, norfloxacin, ciprofloxacin, ofloxacin, levofloxacin, and moxifloxacin (Fig. 1).15–19Ciprofloxacin, which is effective toward Gram-negative bacteria as well as some resistant strains of bacteria, was introduced in the mid-1980s. Further developments led to levofloxacin, an advanced, broad-spectrumantibiotic with strong activity toward a variety of pathogenic Gram-negative and Gram-positive bacteria. Levofloxacin is typically prescribed for a wide range of infections and is the only respiratory fluoroquinolone approved by the US FDA for the treatment of nosocomial pneumonia.
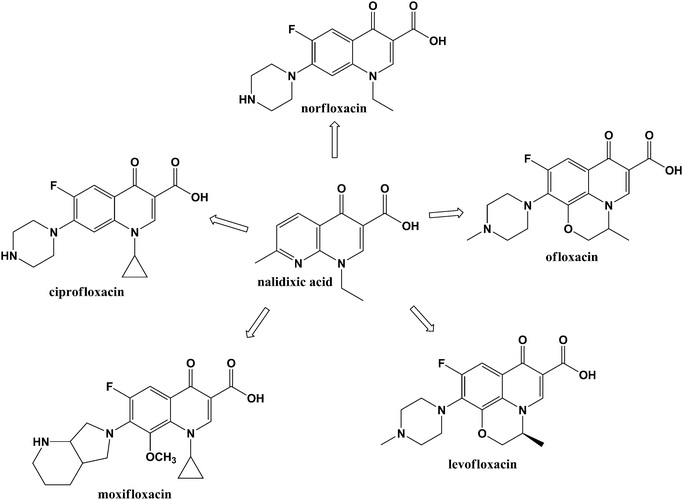 | ||
| Fig. 1 Several antimicrobial fluoroquinolones generated from the parent compound, nalidixic acid. | ||
While fluoroquinolones are an effective class of antibiotics, side effects and limitations are known. Some of the most effective derivatives have been restricted or withdrawn from use due to their toxicity, and there is evidence that bacterial resistance can lead to treatment failure.15,16 There is also great concern that continued widespread use of the most common fluoroquinolones, ciprofloxacin and levofloxacin, will result in the creation of bacterial strains that are resistant to the entire class of fluoroquinolones.
While a variety of polymers have been used for biomedical applications, polysiloxanes and other silicon-based materials, due to their low cytoxicity and desirable physical properties, have been used quite extensively.20–27 Silicones, however, have low surface energy and are hydrophobic leading to poor compatibility with hydrophilic drugs. Hydrophilic modifiers (i.e. channeling agents), such as polyethylene glycol28 or inorganic salts,29 are often necessary for adequate release. The incorporation of these additives decreases the mechanical properties and overall lifetime of the coating or device.
As a means to inhibit the formation of resistant strains of bacteria and eliminate negative side effects associated with systemic treatment, the authors investigated the feasibility of producing polysiloxane-based materials that provide local treatment with levofloxacin. Potential applications for polysiloxane materials that provide local treatment with levofloxacin include coatings for surgical implants,30–33 especially orthopedic devices, which are prone to infection by the Staphylococcus family.
Experimental
Materials
Poly(methylhydro-co-dimethyl)siloxane (HMS-501) (900–1200 g mol−1) and platinum–divinyltetramethyldisiloxane complex in xylene (Karstedt's catalyst) were obtained from Gelest, Inc. (Morrisville, PA). Allyl glycidyl ether, toluene, glacial acetic acid, 33% hydrogen bromide in acetic acid, crystal violet, chloroform, potassiumhydrogen phthalate (KHP), levofloxacin, tetrahydrofuran (THF), methanol, and diethylenetriamine (DETA) were purchased from Sigma-Aldrich (Milwaukee, WI). Intergard 264 was purchased from International Paint LLC (Houston, TX) and DC3140, a silicone coating, was purchased from Dow Corning (Midland, MI). Mueller Hinton (MH) agar was purchased from VWR International (West Chester, PA). The respiratory dye, 2,3,5-triphenyltetrazolium chloride (TTC), was obtained from Amresco (Solon, OH). Phosphate buffered saline (PBS) was obtained from EMD chemicals (Gibbstown, NJ) as a 10× PBS liquid concentrate and diluted to 1× for use in coating preconditioning work by adding appropriate amounts of purified water. All other materials were used as received.Polymer synthesis
An epoxy-functional poly(dimethylsiloxane) copolymer (Ep-PDMS) was synthesized as follows: a magnetic stirring bar, 50 g toluene, 20.0 g (0.148 equivalents Si–H) poly(methylhydro-co-dimethyl)siloxane, 16.9 g (0.148 equivalents allyl) allyl glycidyl ether, and 30 µL platinum–divinyltetramethyldisiloxane complex in xylene were added to a 250 mL round-bottom flask equipped with a condenser and nitrogen purge. The reaction mixture was heated to 80 °C and reaction progress monitored using proton nuclear magnetic spectroscopy (1H NMR) by observing disappearance of the Si–H peak present at 4.8 ppm. Solvent was removed at reduced pressure on a rotary evaporator and the viscous liquid product was collected. The polymer product was characterized by 1H NMR and an epoxy equivalent weight end-grouptitration (ASTM D1652). The polymer produced possessed an epoxy equivalent weight of 272 g eq−1.A polysiloxane copolymer containing both pendant epoxy groups and levofloxacin moieties (levo-Ep-PDMS) was synthesized as follows: 1.8 g (0.0066 equivalents epoxy) Ep-PDMS, 0.09 g (2.5 × 10−4 mol) levofloxacin, 2.0 g methanol, and a magnetic stir bar were added to an 8 mL scintillation vial and the stirred reaction mixture heated with a 70 °C silicone oil bath for 22 h. The product was characterized by NMR, Fourier transform infrared spectroscopy (FT-IR), high performance liquid chromatography (HPLC), gel-permeation chromatography (GPC), and differential scanning calorimetry (DSC).
Coating preparation
A polysiloxane coating solution based on levo-Ep-PDMS was produced by blending 1.9 g (0.0064 equivalents epoxy) of levo-Ep-PDMS in 2.0 g methanol and 138 µL (0.0064 equivalents N–H) DETA in an 8 mL scintillation vial using a Vortex-Genie lab mixer by Scientific Industries (Bohemia, NY). A polysiloxane coating solution containing levofloxacin as an additive was produced by blending 0.025 g (6.9 × 10−5 mol) levofloxacin, 0.51 g (0.0019 equivalents epoxy) Ep-PDMS, 750 µL chloroform, and 42 µL (0.0019 equivalents N–H) DETA in an 8 mL scintillation vial using the Vortex-Genie lab mixer. A PDMS coating solution free of levofloxacin was produced by solution blending 0.5 g (0.0019 equivalents epoxy) Ep-PDMS, 750 µL chloroform, and 42 µL (0.0019 equivalents N–H) DETA in an 8 mL scintillation vial using the Vortex-Genie lab mixer.Specimens for characterization were prepared by depositing coating solutions onto a substrate, allowing the solvent to flash at ambient conditions for 1 h, and curing the coatings at 50 °C for 22 h. The method of deposition and substrate varied with the characterization method used. Details are specified in the forth-coming “Material characterization” section.
Material characterization
NMR spectra were collected using a JEOL ECA400 400 MHz spectrometer equipped with a 24-place autosampler carousel. Polymer samples were measured in deuterated chloroform using 16 scans for protonspectra and a pulse width of 14.6 µs, acquisition time of 2.18 s, pulse angle of 45°, attenuation of 6 dB, pulse time of 7.3 µs, receiver gain of 22, relaxation delay of 4 s, and repetition time of 6.18 s. Carbonspectra were obtained in deuterated chloroform with 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 scans and a pulse width of 10.9 µs, acquisition time of 1.04 s, pulse angle of 30°, attenuation of 9 dB, pulse time of 3.6 µs, receiver gain of 50, relaxation delay of 2 s, and repetition time of 3.04 s. In addition, carbonspectra were run with decoupling and nuclear overhauser effect (NOE) activated at an NOE time of 2 s. Distortionless enhancement by polarization transfer (DEPT) spectra were obtained with decoupling using 17000 scans and a pulse width, acquisition time, and attenuation equivalent to those used for carbonspectra. Irradiation attenuation was 6 dB with a pulse width of 14.6 µs. Receiver gain was set at 60 and selection angle was 135°. Heteronuclear multiple quantum coherence (HMQC) was collected using 112 scans. X-Axis (proton) settings included acquisition time of 0.14 s, attenuation of 6 dB, and pulse width of 14.6 µs. Y-Axis (carbon) settings included acquisition time of 14.97 ms, attenuation of 9 dB, and pulse width of 10.9 µs. Decoupling was active, relaxation delay was 1.5 s, and T1 was 1 µs with a repetition time of 1.64 s. JEOL Delta software and predictions with ChemDraw (CambridgeSoft) software were used to make peak assignments.
000 scans and a pulse width of 10.9 µs, acquisition time of 1.04 s, pulse angle of 30°, attenuation of 9 dB, pulse time of 3.6 µs, receiver gain of 50, relaxation delay of 2 s, and repetition time of 3.04 s. In addition, carbonspectra were run with decoupling and nuclear overhauser effect (NOE) activated at an NOE time of 2 s. Distortionless enhancement by polarization transfer (DEPT) spectra were obtained with decoupling using 17000 scans and a pulse width, acquisition time, and attenuation equivalent to those used for carbonspectra. Irradiation attenuation was 6 dB with a pulse width of 14.6 µs. Receiver gain was set at 60 and selection angle was 135°. Heteronuclear multiple quantum coherence (HMQC) was collected using 112 scans. X-Axis (proton) settings included acquisition time of 0.14 s, attenuation of 6 dB, and pulse width of 14.6 µs. Y-Axis (carbon) settings included acquisition time of 14.97 ms, attenuation of 9 dB, and pulse width of 10.9 µs. Decoupling was active, relaxation delay was 1.5 s, and T1 was 1 µs with a repetition time of 1.64 s. JEOL Delta software and predictions with ChemDraw (CambridgeSoft) software were used to make peak assignments.
A Bruker Optics Vertex 70 FT-IR was used to collect spectra in transmission from samples deposited on a silicon 96-spot array plate. Spectra were analyzed using OPUS software from Bruker.
Polymer molecular weight data were obtained using a Symyx RapidGPC® which consists of a dual-arm liquid handling robot coupled to a temperature-adjustable GPC system using an evaporative light scatteringdetector (Polymer Laboratories ELS 1000) and 2×PLgel mixed-B columns (10 µm particle size). THF was used as the eluent at a flow rate of 2.0 mL min−1 and molecular weights were determined using the aforementioned column and detector at 45 °C by comparing to polystyrene standards.
DSC was performed using a Q2000 from TA Instruments equipped with a liquid nitrogen cooling accessory and 50-place autosampler. After calibration using an indium standard, samples were run in a standard heat–cool–heat protocol from −180 °C to 100 °C at a heating and cooling rate of 10 °C min−1. Tgs were taken as the inflection point of the second heating cycle using TA Universal Analysis software.
Water contact angle was measured on coatings produced by solution casting into a stamped 4″ × 8″ aluminium panel34 using a Symyx integrated contact angle measurement device. The instrument places 10 µL droplets on the coating surface and captures images using a charge-coupled device (CCD) camera for subsequent image analysis and contact angle determination. In addition to static measurements, the instrument was also used to measure water contact angle hysteresis. For water contact angle hysteresis, a 10 µL drop of water is placed on the coating surface and water is added at a constant rate of 0.1 µL s−1 and contact angle measured at 10 s intervals for 1 min. After 1 min, water is removed at 0.1 µL s−1 and contact angle measured at 10 s intervals. Contact angle hysteresis is then calculated by averaging the first three advancing and the last three receding contact angles and subtracting the receding average from the advancing average.
Thermal stability was characterized using a Q500 thermogravimetric analyzer from TA Instruments (New Castle, DE). Using a standard platinum pan, samples were ramped from ambient to 1000 °C at 20 °C min−1. Nitrogen was used as both the balance gas (40 mL min−1) and the sample gas (60 mL min−1) with an air-cool time of 25 min between samples.
Optical images were collected on coatings deposited on glass cover slips using an Olympus BX61 epifluorescence microscope, and images were collected with a DP70 12.5 million pixel digital camera. Fluorescence images were obtained using fluorescence excitation with a green filter for 20 ms.
The antimicrobial activity of coatings on aluminium discs pre-coated with an epoxy primer (Intergard 264) and silicone-based tiecoat (Dow Corning 3140) was determined by placing the coated discs in 20 mL scintillation vials containing 5 mL of 1× PBS for 24 h at ambient conditions. The 1× PBS solution was removed and the scintillation vials, containing the coating discs, were placed in a desiccator for 4 h to facilitate drying of the coating surfaces. The coating discs were then inverted and placed on a MH agar plate inoculated with ∼107cells mL−1 of the Gram-negative bacterium, Escherichia coliATCC 12435. TTC was incorporated into the MH agar (70 mg L−1) to facilitate visualization of bacterial growth on the coating surfaces (red color). Agar plates with inverted coating discs were incubated for 24 h at 37 °C and examined for both zones of growth inhibition and inhibition of growth on the coating surfaces. The coating discs were then removed from the MH agar plates and cleaned by gently wiping the coating surfaces with a moist Kimwipe®. The cleaned coating discs were placed into new 20 mL scintillation vials containing 5 mL of fresh 1× PBS for an additional 6 days (total of 7 days immersion) at ambient laboratory conditions and evaluated for antimicrobial activity using the agar plating method described above. This process was repeated several times to determine the antimicrobial activity of the coating discs to 81 days of total immersion in 1× PBS.
Results and discussion
Polymer synthesis and characterization
As shown in Fig. 2, the synthesis of levo-Ep-PDMS was a two step process in which glycidyl ethergroups were first grafted to a poly(methylhydro-co-dimethyl)siloxane copolymer using hydrosilylation chemistry to produce Ep-PDMS. A portion of the epoxides present on Ep-PDMS was reacted with levofloxacin to produce levo-Ep-PDMS. NMR was used to characterize levo-Ep-PDMS as well as the polymer intermediate, Ep-PDMS. Fig. 3 displays NMR spectra obtained for Ep-PDMS along with peak assignments. Peak assignments were determined using the four spectral techniques shown in Fig. 3. Using integration of proton peaks, the number of protons corresponding to each peak was determined. Carbonspectra provided the carbon peak information. DEPT135 was used to distinguish primary, secondary, tertiary and quaternary substituted carbon atoms. This technique provides spectra in which methyl (primary) and methine (tertiary) peaks orient in one direction, while methylene (secondary) carbon peaks orient in the opposite direction. Tetra-substituted (quaternary) carbon peaks do not appear in the DEPT135 spectrum. By comparing to a carbonspectrum, assignments are easily made. HMQC is a two-dimensional technique that correlates proton and carbon peak information. A peak is observed when protons and carbons are associated with one another. From HMQCspectra, complex protonspectra can be assigned through correlation with the appropriate carbon atoms. In the 1H NMR, the presence of the peaks corresponding to the protons of the methylene group (“o” in Fig. 3) and the lack of a peak at 4.8 ppm corresponding to silicon hydrideprotons demonstrate successful grafting of glycidyl ethergroups to the polysiloxane backbone. The formation of pendant levofloxacingroups by reaction of carboxylic acidgroups of levofloxacin with glycidyl groups of Ep-PDMS was demonstrated by the observation of peaks in both the 1H NMR and 13C NMR spectra that correspond to the isopropyl group generated by ring opening of the glycidyl ethergroups of Ep-PDMS (Fig. 4). Levofloxacin has been previously characterized by NMR.35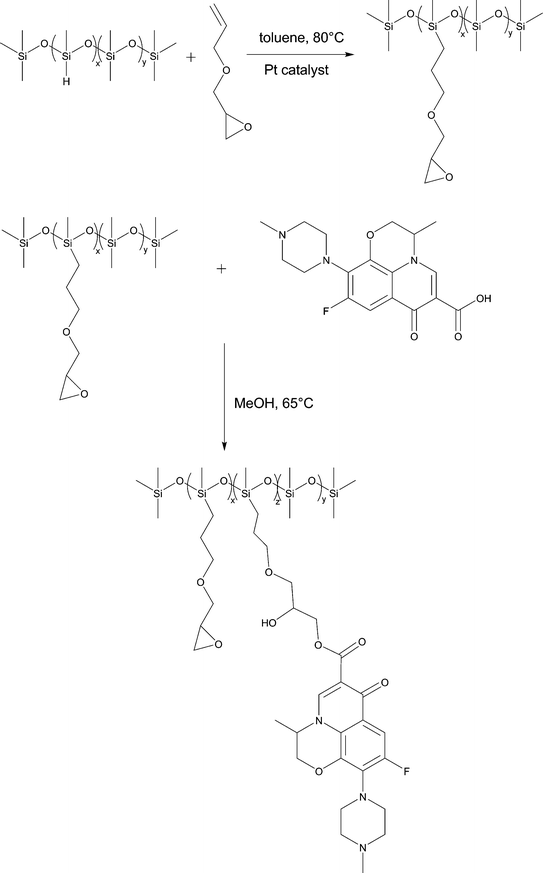 | ||
| Fig. 2 Synthetic scheme used to produce epoxy-functional levofloxacin-containing polysiloxane copolymers. | ||
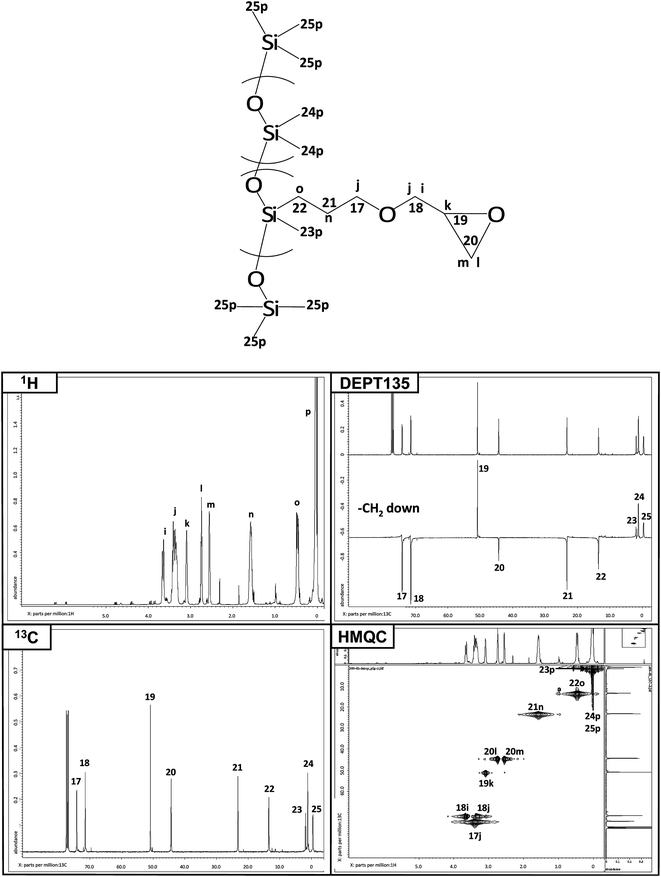 | ||
| Fig. 3 NMR characterization of Ep-PDMS. | ||
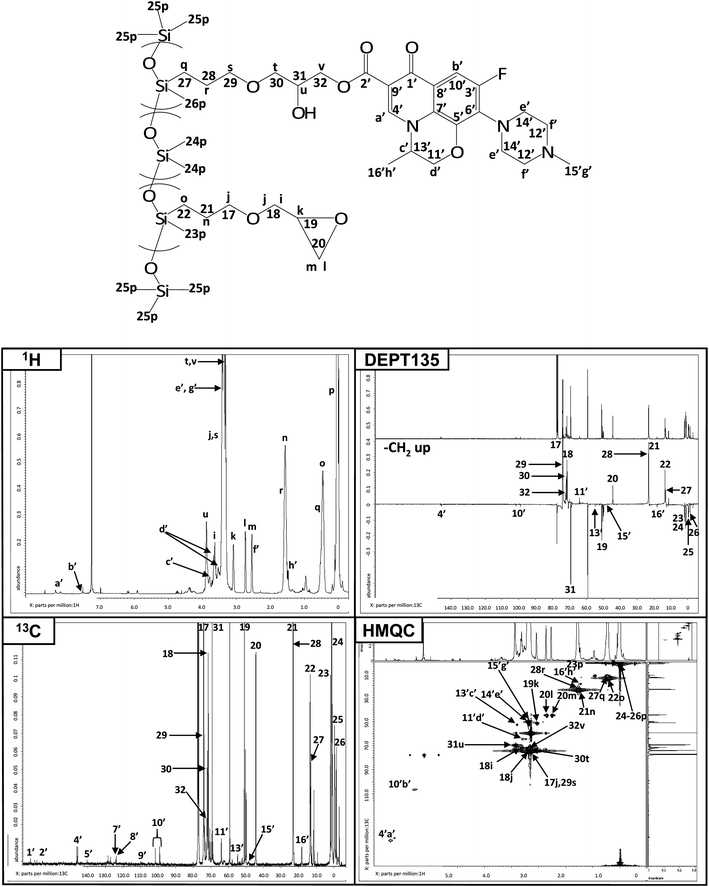 | ||
| Fig. 4 NMR characterization of levo-Ep-PDMS. | ||
The reaction of levofloxacin with Ep-PDMS was also characterized using FT-IR (Fig. 5). Levofloxacin has been characterized previously by FT-IR.36 Briefly, bands in the levofloxacinspectrum are as follows: 3268 cm−1 (OH), 2959–2803 cm−1 (CH3), 1722 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O acid), 1622 cm−1 (CO ring). Bands for Ep-PDMS are as follows: 2959–2803 cm−1 (–CH3, –CH2–), 1259 cm−1 (Si–CH3), 1193 cm−1 (Si–CH2–), 1090 cm−1 (–Si(CH3)2–O–), 1026 cm−1 (–Si(CH3)2–O–), 911 cm−1 (epoxy ring), 846 cm−1 (Si–CH3), and 800 cm−1 (Si–CH3). For the reaction product, levo-Ep-PDMS, an increase in the OH band at 3411 cm−1 can be seen which was due to the formation of the secondary hydroxylgroup resulting from the epoxy ring-opening reaction. In addition, evidence for the formation of the ester tether was obtained by comparing the carbonyl region (1722 cm−1 to 1592 cm−1) of spectra obtained before and after reaction. The reaction resulted in a large reduction in the band for the carboxylic acid carbonyl of levofloxacin at 1722 cm−1. Also observed was a relative increase in the carbonyl band at 1622 cm−1 and the formation of a new carbonyl band at 1592 cm−1. These changes in the carbonyl bands suggest the formation of the ester tether via epoxy ring-opening by the levofloxacin carboxylic acid group. Although significantly reduced in intensity, the existence of the carboxylic acid carbonyl band at 1722 cm−1 indicated the presence of some unreacted levofloxacin in the polymer sample.
O acid), 1622 cm−1 (CO ring). Bands for Ep-PDMS are as follows: 2959–2803 cm−1 (–CH3, –CH2–), 1259 cm−1 (Si–CH3), 1193 cm−1 (Si–CH2–), 1090 cm−1 (–Si(CH3)2–O–), 1026 cm−1 (–Si(CH3)2–O–), 911 cm−1 (epoxy ring), 846 cm−1 (Si–CH3), and 800 cm−1 (Si–CH3). For the reaction product, levo-Ep-PDMS, an increase in the OH band at 3411 cm−1 can be seen which was due to the formation of the secondary hydroxylgroup resulting from the epoxy ring-opening reaction. In addition, evidence for the formation of the ester tether was obtained by comparing the carbonyl region (1722 cm−1 to 1592 cm−1) of spectra obtained before and after reaction. The reaction resulted in a large reduction in the band for the carboxylic acid carbonyl of levofloxacin at 1722 cm−1. Also observed was a relative increase in the carbonyl band at 1622 cm−1 and the formation of a new carbonyl band at 1592 cm−1. These changes in the carbonyl bands suggest the formation of the ester tether via epoxy ring-opening by the levofloxacin carboxylic acid group. Although significantly reduced in intensity, the existence of the carboxylic acid carbonyl band at 1722 cm−1 indicated the presence of some unreacted levofloxacin in the polymer sample.
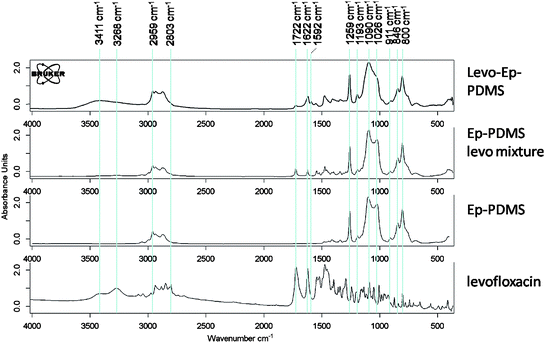 | ||
| Fig. 5 FT-IR spectra used to characterize the levofloxacin tethering reaction: levofloxacin; Ep-PDMS; physical mixture of Ep-PDMS with levofloxacin; and levo-Ep-PDMS. | ||
GPC was also used to characterize the reaction of levofloxacin with Ep-PDMS. Fig. 6 compares GPCchromatograms for Ep-PDMS, levofloxacin, a physical blend of Ep-PDMS and levofloxacin corresponding to the reaction mixture used to prepare levo-Ep-PDMS, and the reaction product, levo-Ep-PDMS. The chromatogram obtained for levo-Ep-PDMS lacks the peak centered at 525 s which is associated with Ep-PDMS. In addition, the single peak obtained for levo-Ep-PDMS was shifted to higher retention time and the molecular weight distribution was narrowed as compared to Ep-PDMS. These results provide further evidence that levofloxacin was successfully tethered to Ep-PDMS. The reduction in retention time and narrowing of the molecular weight distribution obtained by tethering of levofloxacin to Ep-PDMS suggest that attachment of the levofloxacin molecule to the polysiloxane as a side chain results in intramolecular interactions in THF that reduce the hydrodynamic volume of the polymer.
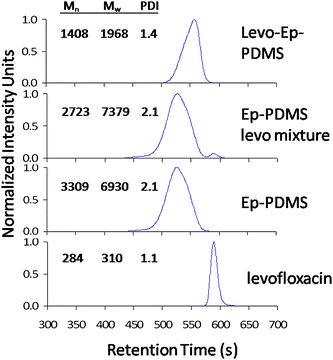 | ||
| Fig. 6 GPC chromatograms showing molecular weight distribution differences between: levofloxacin; Ep-PDMS; physical mixture of Ep-PDMS with levofloxacin; and levo-Ep-PDMS. | ||
The thermal properties of Ep-PDMS and levo-Ep-PDMS were determined using DSC and TGA. As shown in Fig. 7, Ep-PDMS was an amorphous polymer exhibiting a single Tg at −89 °C. The reaction of Ep-PDMS with levofloxacin to produce levo-Ep-PDMS resulted in an 18 °C shift in Tg consistent with the bulky levofloxacin moiety significantly reducing segment mobility of the polysiloxane polymer backbone.
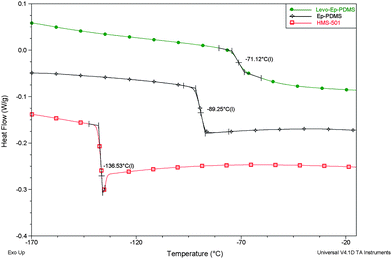 | ||
| Fig. 7 DSC thermograms for HMS-501, Ep-PDMS, and levo-Ep-PDMS. | ||
Fig. 8 provides a comparison of the thermal stability of Ep-PDMS, levofloxacin, and levo-Ep-PDMS. Levo-Ep-PDMS shows approximately a 5% weight loss between 140 °C and the onset of the primary decomposition process at 325 °C. This weight loss is most likely due to the loss of absorbed water resulting from the hydrophilic nature of the levofloxacin moieties.
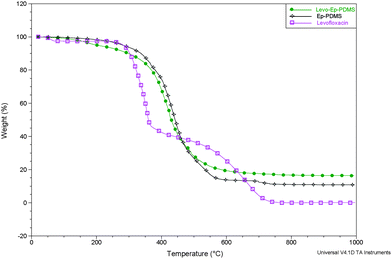 | ||
| Fig. 8 A comparison of thermal stability by TGA of levo-Ep-PDMS, Ep-PDMS, and levofloxacin. | ||
Coating characterization results
A surface coating, which will be referred to as “tethered,” was prepared from levo-Ep-PDMS by solution blending with DETA which served as a crosslinker for the pendant epoxy groups. For comparison purposes, two other coatings were produced which will be referred to as “control” and “doped.” The control coating was Ep-PDMS crosslinked with DETA while the doped coating was basically the control with 5 wt% levofloxacin as an additive in the coating, i.e. the levofloxacin was not chemically attached to the coating matrix. Fig. 9 displays images of the three coatings deposited on glass cover slips. The opacity observed for the doped coating clearly indicated that the levofloxacin was insoluble in the polysiloxane matrix. Covalently linking, i.e. tethering, the levofloxacin to the polymer matrix prevented macroscopic phase separation as indicated by the transparent coating produced.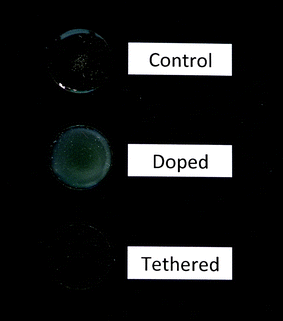 | ||
| Fig. 9 Images of the three crosslinked coatings produced. “Control” was produced by crosslinking Ep-PDMS with DETA. “Tethered” was produced by crosslinking levo-Ep-PDMS with DETA. “Doped” was produced by solution blending levofloxacin, Ep-PDMS, and DETA. | ||
Further characterization of the morphology of these coatings was conducted using both optical and fluorescence microscopy. Since levofloxacin fluoresces strongly, tethered and doped samples were produced using 1.0 wt% levofloxacin as opposed to 5.0 wt% to facilitate morphological characterization. As shown in Fig. 10, the levofloxacin moieties were uniformly distributed throughout the tethered coating while the fluorescence for the doped coating was non-uniform due to insolubility or limited solubility of the levofloxacin in the coating matrix.
 | ||
| Fig. 10 Images from optical (left) and fluorescence (right) microscopy of coatings made on glass cover slips containing 1 wt% “doped” (a and c) and “tethered” (b and d) levofloxacin. | ||
As shown in Fig. 11, the Tg of the control coating and doped coating was the same (−85 °C) while that of the tethered coating was 40 °C higher due to limitations on polymer chain segmental mobility induced by the bulky levofloxacin moieties. The equivalence of the Tg obtained for both the control coating and doped coating indicated that reaction of levofloxacin with the epoxy groups of Ep-PDMS did not occur to any significant extent during curing of the coating at 50 °C. While the Tg of the tethered coating was significantly higher than that of a typical crosslinked PDMS elastomer, it was still well below room temperature and, as a result, exhibited the rubbery character desired for many biomedical applications.
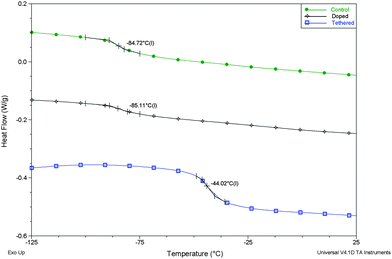 | ||
| Fig. 11 DSC thermograms of the coatings produced. | ||
The high polymer backbone flexibility of PDMS enables the production of a regular array of methyl groups to be oriented outward to the air interface resulting in a highly hydrophobic surface.37 Since surface wettability and water egress into the coating will strongly affect antimicrobial activity, water contact angle and water contact angle hysteresis were measured. As shown in Table 1, both the control coating and the doped coating exhibited the high water contact angle typically observed for PDMS, while the tethered coating displayed a significantly lower water contact angle. The similarity between the contact angle obtained for the control and doped coatings indicated that the composition of the doped coating at the very outer surface was essentially polysiloxane methyl groups. Considering the insolubility of the levofloxacin in the coating matrix, it was not a surprise that the composition of the coating surface would be largely comprised of the lower surface energy polysiloxane matrix. Since the levofloxacin moieties present in the tethered coating are covalently bound to the polysiloxane matrix, phase separation is limited and polymer backbone mobility hindered. Due to these factors, the chemical composition of the coating surface may include some fraction of levofloxacin moieties resulting in the lower water contact angle.
| Water contact angle/° | Contact angle hysteresis | |
|---|---|---|
| Control | 90.48 ± 1.11 | 2.51 |
| Doped | 94.20 ± 2.68 | 13.01 |
| Tethered | 84.80 ± 1.38 | 12.47 |
Contact angle hysteresis (CAH) is a general indicator of surface chemical or morphological stability.38,39 In general, CAH can be used as an indication of the degree of surface instability resulting from wetting of the surface. The control coating had a low CAH, indicative, in this case, of a surface stable to water-induced rearrangement. Both the doped and tethered coatings, on the other hand, showed a large relative CAH, indicative of surface instability.
Antimicrobial characterization results
Antimicrobial activity of the coatings was determined as a function of immersion time in PBS using agar plates inoculated with E. coli. As shown in Fig. 12, the control coating showed extensive microbial growth on the surface of the coating and no evidence of a zone of inhibition. In contrast, both the doped and tethered coatings showed antimicrobial activity on the surface of the coating and a relatively large zone of growth inhibition. Interestingly, the zone of growth inhibition for the tethered coating was initially much larger than that for the doped coating indicating greater levofloxacin release from the tethered coating. This result may be due to differences in coating morphology and levofloxacin dispersion as well as the presence of some “free,” i.e. unreacted, levofloxacin in the tethered coating. The optical and fluorescence microscopy results showed that the levofloxacin phase separated from the polysiloxane matrix for the doped coating while no evidence of macroscopic phase separation was observed for the tethered coating. Considering the insolubility of levofloxacin in the polysiloxane matrix and the low surface energy of polysiloxanes, the smaller zone of growth inhibition exhibited by the doped coating may be due to the generation of a coating surface morphology consisting of a largely polysiloxane-rich composition at the coating-air interface that is much lower in levofloxacin than the bulk of the coating. Since the levofloxacin is covalently bound to the polysiloxane matrix for the tethered coating, levofloxacin moieties must necessarily be in the vicinity of the coating-air interface thereby facilitating release from the coating.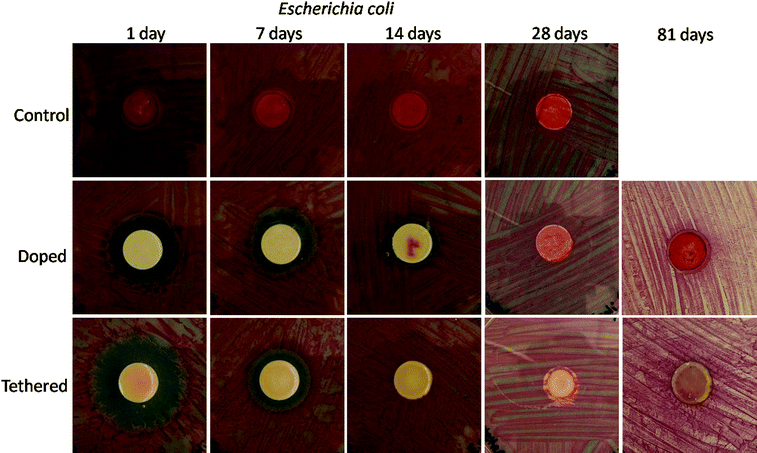 | ||
| Fig. 12 Images of coated specimens preconditioned in PBS for various time periods and tested for antimicrobial activity toward E. coli using an agar plating method. The “darkness” observed on the surface of the 81 day tethered disc is a result of repeated measurement of the same samples and not due to growth of E. coli on the surface. | ||
After 28 days of immersion in PBS, the doped coating displayed no antimicrobial activity while the tethered coating showed antimicrobial activity exclusively at the coating surface. This result suggests that the zone of growth inhibition observed for the tethered coating before immersion was most likely due to leaching of residual, unreacted levofloxacin from the coating. After sufficient leaching of free levofloxacin from the coating, only tethered levofloxacin moieties remained thus eliminating the observation of a zone of growth inhibition. The long-term activity is due to the slow release of levofloxacin through a hydrolytic mechanism. The presence of some “free” or unreacted levofloxacin in the tethered coating is consistent with the FT-IR results discussed earlier in this document.
Conclusion
The broad-spectrumantimicrobial agent, levofloxacin, was successfully tethered to Ep-PDMS by ring-opening pendant epoxides to produce an ester-functional tether. Attachment of levofloxacin to the polysiloxane resulted in a significant increase in Tg. The levo-Ep-PDMS was then used to produce a crosslinked coating by reacting pendant epoxides with DETA. Unlike the doped coating, the tethered coating was a one-phase material in which the levofloxacin moieties were uniformly dispersed throughout the coating. With regard to antimicrobial activity, the tethered coating allowed for a greater initial release of levofloxacin and prolonged activity toward E. coli when compared to the doped coating. The greater initial release of levofloxacin from the tethered coating was attributed to the more uniform distribution of levofloxacin as well as to the presence of some “free,” i.e. unreacted, levofloxacin. These results suggest that tethering levofloxacin to a polysiloxane matrix may result in the production of new surface coatings to combat implanted device-related infection.Acknowledgements
The authors acknowledge the Office of Naval Research under grant N00014-07-1-1099. Special thanks to Kenneth Johnson for assistance in statistical design analysis pertaining to these coatings.References
- R. A. Deyo, A. Nachemson and S. K. Mirza, N. Engl. J. Med., 2004, 350, 722–726 CrossRef CAS.
- R. O. Darouiche, N. Engl. J. Med., 2004, 350, 1422–1429 CrossRef CAS.
- O. Darouiche Rabih, Clin. Infect. Dis., 2003, 36, 1284–1289 CrossRef CAS.
- M. Lucke, B. Wildemann, S. Sadoni, C. Surke, R. Schiller, A. Stemberger, M. Raschke, N. P. Haas and G. Schmidmaier, Bone, 2005, 36, 770–778 CrossRef CAS.
- S. Conti, L. Polonelli, R. Frazzi, M. Artusi, R. Bettini, D. Cocconi and P. Colombo, Curr. Pharm. Biotechnol., 2000, 1, 313–323 CAS.
- S. Kumar and A. Ali, Indian J. Hosp. Pharm., 1995, 32, 129–133 Search PubMed.
- A. Kydonieus, Treatise on Controlled Drug Delivery—Fundamentals, Optimisation, Applications, Marcel Dekker, New York, 1992 Search PubMed.
- Y. Loo and K. W. Leong, Biomaterials for drug and gene delivery, An Introduction to Biomaterials (Biomedical Engineering), ed. S. A. Guelcher and J. O. Hollinger, CRC Press, Boca Raton, FL, 2005, pp. 342–368 Search PubMed.
- L. Masaro and X. X. Zhu, Prog. Polym. Sci., 1999, 24, 731–775 CrossRef CAS.
- M. V. S. Varma, A. M. Kaushal, A. Garg and S. Garg, Am. J. Drug Delivery, 2004, 2, 43–57 Search PubMed.
- C. von Eiff, B. Jansen, W. Kohnen and K. Becker, Drugs, 2005, 65, 179–214 CrossRef CAS.
- C. Von Eiff, W. Kohnen, K. Becker and B. Jansen, Int. J. Artif. Organs, 2005, 28, 1146–1156 Search PubMed.
- I. Francolini, G. Donelli and P. Stoodley, Rev. Environ. Sci. Bio/Technol., 2003, 2, 307–319 Search PubMed.
- E. M. Hetrick and M. H. Schoenfisch, Chem. Soc. Rev., 2006, 35, 780–789 RSC.
- F. Van Bambeke, J. M. Michot, J. Van Eldere and P. M. Tulkens, Clin. Microbiol. Infect., 2005, 11, 256–280 Search PubMed.
- P. Ball, Curr. Ther. Res., 2003, 64, 646–661 CrossRef CAS.
- K. F. Croom and K. L. Goa, Drugs, 2003, 63, 2769–2802 CrossRef CAS.
- M. Hurst, H. M. Lamb, L. J. Scott and D. P. Figgitt, Drugs, 2002, 62, 2127–2167 CrossRef CAS.
- A. J. Schaeffer, Am. J. Med., 2002, 113, 45S–54S CAS.
- F. Abbasi, H. Mirzadeh and A.-A. Katbab, Polym. Int., 2001, 50, 1279–1287 CrossRef CAS.
- ACS Symposium Series: Science and Technology of Silicones and Silicone-Modified Materials, ed. S. J. Clarson, J. J. Fitzgerald, M. J. Owen, S. D. Smith and M. E. Van Dyke, Oxford University Press, New York, NY, 2007, vol. 64, p. 400 Search PubMed.
- R. A. Compton, J. Long-Term Eff. Med. Implants, 1997, 7, 29–54 CAS.
- Silicon Containing Polymers: The Science and Technology of Their Synthesis and Applications, ed. R. G. Jones, W. Ando and J. Chojnowski, Kluwer Academic Publishers, Norwell, MA, 2000, p. 768 Search PubMed.
- C. R. McMillin, Rubber Chem. Technol., 1994, 67, 417–446 CAS.
- C. R. McMillin, Rubber Chem. Technol., 2006, 79, 500–519 CAS.
- Biomaterials Science, An Introduction to Materials in Medicine, ed. B. D. Ratner, A. S. Hoffman, F. J. Schoen and J. E. Lemons, Elsevier Academic Press, London, 2nd edn, 2004, p. 851 Search PubMed.
- R. Yoda, J. Biomater. Sci., Polym. Ed., 1998, 9, 561–626 CrossRef CAS.
- J. Schulze Nahrup, Z. M. Gao, J. E. Mark and A. Sakr, Int. J. Pharm., 2004, 270, 199–208 CrossRef.
- G. Di Colo, Controlled drug release from implantable matrixes based on hydrophobic polymers, in Proceedings and Program of the 18th International Symposium on Controlled Release of Bioactive Materials, ed. I. W. Kellaway, Controlled Release Society, Deerfield, IL, 1991, pp. 317–318 Search PubMed.
- H. S. El-Zaim and J. P. Heggers, Silicones for pharmaceutical and biomedical applications, in Polymeric Biomaterials, ed. S. Dumitriu, Marcel Dekker, Inc., New York, NY, 2nd edn, 2002, pp. 79–90 Search PubMed.
- B. Gottenbos, H. C. van der Mei, F. Klatter, P. Nieuwenhuis and H. J. Busscher, Biomaterials, 2002, 23, 1417–1423 CrossRef CAS.
- T. J. Laing, D. Schottenfeld, J. V. LaceyJr, B. W. Gillespie, D. H. Garabrant, B. C. Cooper, S. G. Heeringa, K. H. Alcser and M. D. Mayes, Am. J. Epidemiol., 2001, 154, 610–617 CrossRef CAS.
- J. J. H. Oosterhof, K. J. D. A. Buijssen, H. J. Busscher, B. F. A. M. van der Laan and H. C. van der Mei, Appl. Environ. Microbiol., 2006, 72, 3673–3677 CrossRef CAS.
- B. J. Chisholm, S. J. Stafslien, D. A. Christianson, C. Gallagher-Lein, J. W. Daniels, C. Rafferty, L. Vander Wal and D. C. Webster, Appl. Surf. Sci., 2007, 254, 692–698 CrossRef CAS.
- G. Fardella, P. Barbetti, I. Chiappini and G. Grandolini, Int. J. Pharm., 1995, 121, 123–127 CrossRef CAS.
- S. Sagdinc and S. Bayari, THEOCHEM, 2004, 668, 93–99 CrossRef CAS.
- M. J. Owen, in Surface Properties and Applications, ed. R. G. Jones, W. Ando and J. Chojnowski, Kluwer Academic Publishers, Dordrecht, 2000 Search PubMed.
- P. Majumdar, S. Stafslien, J. Daniels and D. C. Webster, J. Coat. Technol. Res., 2007, 4, 131–138 Search PubMed.
- J. H. Wang, P. M. Claesson, J. L. Parker and H. Yasuda, Langmuir, 1994, 10, 3887–3897 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |