Controlled room temperature ROP of L-lactide by ICl3: a simple halogen-bonding catalyst
Olivier
Coulembier
*,
Franck
Meyer
and
Philippe
Dubois
Center of Innovation and Research in Materials and Polymers (CIRMAP), Laboratory of Polymeric and Composite Materials, University of Mons, 23 Place du Parc, B-7000, Mons, Belgium. E-mail: olivier.coulembier@umons.ac.be; Fax: +0032 (0)65 373484; Tel: +0032 (0)65 373480
First published on 18th February 2010
Abstract
Halogen-bonding ROPactivation of L-lactide monomer by ICl3 at room temperature leads to well-controlled poly(L-lactide)s of predictable molecular weights and narrow molecular weight distributions.
Halogen bonding (XB) is commonly defined as a charge-transfer interaction involving halogens as electrophilic sites.1 The attractive nature of XB is highly selective and directional, which makes them new and reliable items in the array of weak interactions. Thus, the deliberate crystal engineering relying on halogen bonded molecular units has demonstrated a remarkable ability in the elaboration of complicated supramolecular structures.2 Furthermore, recent studies have reported the design of halogen bonded functional materials with applications in solid state synthesis, non linear optics, liquid crystals, imprinted polymers and very recently, XB has been also considered as a rational approach in drug design.3
Even if iodine-based structures have been used to promote polymerization of styrene and vinylether derivatives, (tetrahydro)furans and several cyclophanes,4 to the best of our knowledge, very limited interest has been devoted to use of halogens as catalysts in polymerization reactions. As far as the self-assembling process is concerned, the formation of supramolecular complexes supported by X⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C halogen bonds is not a common feature5 and ring-opening polymerization (ROP) of (di)lactones has been attempted.
C halogen bonds is not a common feature5 and ring-opening polymerization (ROP) of (di)lactones has been attempted.
Herein, we demonstrate the possibility of using iodine trichloride (ICl3) to promote the ROP of L-lactide (LA) in a selective and controlled manner at room temperature. Quite interestingly, the simple commercial ICl3 proved to activate both the monomer and the alcohol initiator, presenting a simplicity that some of the recent non-organocatalytic systems do not have due to the use of co-catalysts or their simple non-commercially availability.6 Electrophilic activation of carbonyl groups with Lewis acids is a well-established method for enhancing their reactivity and selectivity toward nucleophilic addition.7 Here the halogen bonding capability of the ICl3catalyst was first assessed by infra-red spectroscopy. The monitoring of a CHCl3 solution of LA under ICl3 addition results in a continuous shift of the νCO maximum frequency band from ca. 1759 to 1772 cm−1, consistent with an electron donation from the carbonyl group toward the iodine (Fig. 1).8 The CO vibrational signal shifts mainly during the initial titration step with a maximum displacement observed for a catalyst-to-LA molar ratio of ∼3. This result suggests that the carbonyloxygen atoms may interact with at least one ICl3viaXB when an excess of ICl3 is used. As shown by 13C NMR (Fig. 2), the addition of one molar equivalent of ICl3 with respect to LA monomer results in a clear downfield shift split of the carbonyl group present at 168.49 ppm (Δ ≈ 1.8 ppm). Methyl and methine carbons signals are also split and shifted to lower field, attesting for the strong XB interaction between the iodine and oxygen atoms. Since two sets of signals (α, β, γ and α′, β′, γ′) are present at r.t., we might expect a really slow interaction exchange between both LA and ICl3 at room temperature.
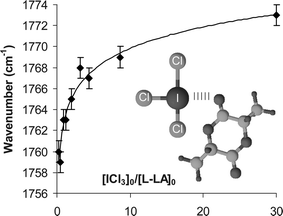 | ||
| Fig. 1 FT-IR wavenumber shifts observed by titration of LA with ICl3 (carbonyl absorption, solvent = CHCl3). | ||
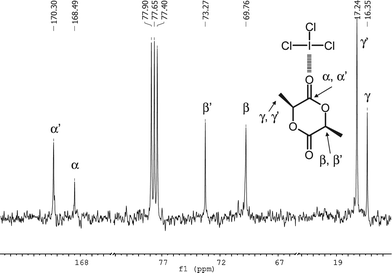 | ||
| Fig. 2 13C NMR spectrum of a LA/ICl3 mixture (1/1) in CDCl3. | ||
Since alcohols are used as nucleophilic intiatiors in ROP of lactides, a possible interaction between ICl3 and a representative alkyl primary alcohol (11-bromo-1-undecanol, 11-BU) was subsequently investigated by using 1H NMR spectroscopy. When 11-BU was added to 1 molar equivalent of ICl3, the hydroxylproton (Hγ) was significantly affected as indicated by a large downfield shift of the OH resonance (Δ ≈ 7.4 ppm), consistent with OH⋯Clhydrogen bonds (HB). This strong electron-withdrawing effect was also observed for methyleneprotons in α (Hϕ) and β-positions (Hε) of the OHgroup as characterized by shifts of 0.25 and 0.2 ppm, respectively (Fig. 3).
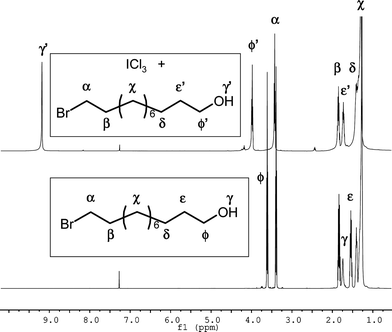 | ||
| Fig. 3 1H NMR spectra of 11-BU (bottom) and an 11-BU/ICl3 mixture (1/1) (top) in CDCl3. | ||
Combined multinuclear NMR and FT-IR spectroscopy results suggest that both monomer and initiator interact with the ICl3catalyst through strong intermolecular XB and HB interactions, respectively. For that reason, ICl3 was used as a possible catalyst for the ROP of LA using 11-BU as the initiator (Scheme 1).
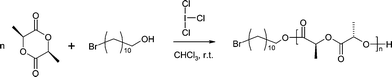 | ||
| Scheme 1 | ||
The polymerization of LA was carried out in CHCl3 at r.t. using 11-BU as initiator for different monomer-to-initiator-to-catalyst ratios ([LA]0 = 2.3 M, Table 1).
| Entry | [LA]0/[I]0/[C]0 | Time/h | Conv (%)b | Mnth/g mol−1 | Mnexpc/g mol−1 | PDIc |
|---|---|---|---|---|---|---|
| a Conditions: [M]0 = 2.3 M, solvent: CHCl3, r.t. b Determined by 1H NMR spectroscopy and/or gravimetry. c Molecular weight and polydispersity index as determined by gel permeation chromatography in THF after appropriate Mark-Houwink-Sakurada parameters application. | ||||||
| 1 | 50/1/1 | 23 | 26 | 1900 | 1300 | 1.1 |
| 2 | 50/1/1 | 71 | 35 | 2500 | 2500 | 1.3 |
| 3 | 174/1/3 | 66 | 22 | 5500 | 4800 | 1.4 |
| 4 | 174/1/10 | 23 | 36 | 9000 | 9800 | 1.2 |
| 5 | 174/1/10 | 44 | 64 | 16000 | 14000 | 1.3 |
| 6 | 174/1/15 | 44 | 86 | 21600 | 21300 | 1.4 |
Due to the slow intermolecular exchange between ICl3 and LA (cf.Fig. 2), the polymerization occurs slowly at room temperature when an equimolar quantity of ICl3 is used regarding the initiator (Table 1, entries 1 and 2). If 71 h are required to lead to poly(L-lactide) (PLA) chains with a polymerization degree (DP) of 18, excess of catalyst gradually increases the propagation kinetics and molecular weights up to 21 kg mol−1 are obtained within 44 h (entry 6). Comparison between the PLA molecular weights (Mnexp) and those calculated gives a linear fit up to 21,000 g mol−1, consistent with a controlled polymerization. Gel permeation chromatograms show a Gaussian distribution of molecular weights for each sample, with polydispersity indexes (PDIs) varying from 1.1 to 1.4.
A plausible polymerization pathway is through a double activation of both initiator and monomer mechanism in which initiation occurs when a pronounced nucleophilic alcohol reacts with a LA-ICl3 complex to form the mono-adduct, and the α-chain end of the growing PLA bears an ester functionality derived from the alcohol (eqn (1)).9
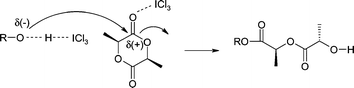 | (1) |
Polymerization proceeds when the terminal ω-hydroxylgroup (activated by ICl3) acts as a nucleophile to facilitate further chain extension. It is important to note that the number of ICl3 molecules necessary to activate both monomer and the alcohol initiator is not known precisely and either one or at least two molecules might be involved into the activation process.
While 1H NMR analysis of PLA consistently identifies both expected end-groups (Fig. 4), electrospray ionization mass spectrometry measurement (ESI-MS) shows almost exclusively peaks corresponding to the BrC11H22O(C(O)CH(CH3)O)nH population doped with Na+ (Fig. 5). Simply and doubly charged most intensive population signals agree perfectly well with the expected macromolecular structures and GPC data of a representative PLA (Table 1, entry 1, Mnexp = 1300 g mol−1). In Fig. 6, one of the signals from the Fig. 5 population, namely BrC11H22O(C(O)CH(CH3)O)16H, Na+, is expanded. The computed isotopic distribution for the same signal reproduces the experimental signal almost perfectly.
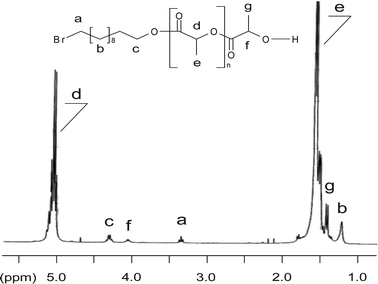 | ||
| Fig. 4 1H NMR analysis of a poly(L-lactide) as obtained by LA ROP from 11-BU and ICl3 as initiator and catalyst, respectively. | ||
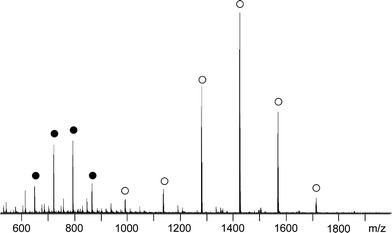 | ||
| Fig. 5 ESI mass spectrum of PLA (Table 1, entry 1) simply (○) and doubly (●) charged (observed charges: Na+). | ||
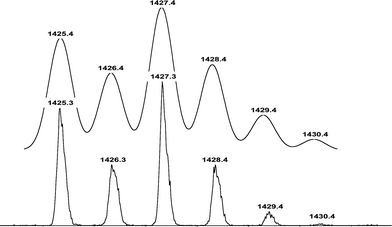 | ||
| Fig. 6 Comparison of the 1425–1431 m/z fragment of the ESI-MSspectrum in Fig. 5 (bottom) with the isotopic distribution computed for the following species: BrC11H22O(C(O)CH(CH3)O)16H, Na+ (top). | ||
In conclusion, the ability of ICl3 to activate electrophilic substrates through halogen bonding was demonstrated by NMR and FT-IR spectroscopies. The catalytic activity of the XB donor towards ROP produced narrowly dispersed PLA of predictable molecular weights. Elevated temperature experiments are currently under study to evaluate a possible kinetic improvement and will be the topic of a forthcoming paper.
Experimental part
Materials
L-Lactide (GALACTIC, Belgium) was recrystallized from dried toluene and stored in a glove box under dry nitrogen atmosphere before use. 11-Bromo-1-undecanol (Aldrich, 99%) was dried by three successive azeotropic distillation by addition of tetrahydrofuran and stored in a glove box under dry nitrogen atmosphere before use. Iodine trichloride (Aldrich, 95%) was used without any purification. Tetrahydrofuran (THF) and chloroform (CHCl3) solvents were dried using a MBraun SolventPurification System (model MB-SPS 800) equipped with alumina drying columns.Characterizations
FT-IR analyses were performed in a solid state by using a Fourier Transform Infra-Red Bruker Tensor 27 Spectrometer (6000–600 cm−1) equipped with a Miracle micro-ATR from Pike. Lactide-ICl3 mixtures were prepared by dissolving LA and ICl3 in chloroform. The solution was slowly evaporated and the fine mixture analyzed after 5 min. 1H and 13C NMR spectra were recorded using both a Bruker AMX-300 and Bruker AMX-500 apparatus at r.t. in CDCl3 (30 mg/0.6 ml). The molecular mass measurements as well as the end-group determinations were performed on a Waters QToF2 mass spectrometer equipped with an orthogonal electrospray (ESI) source (Z-spray) operated in positive ion mode. PLA was dissolved in acetonitrile in order to approximately achieve 10−4 M concentrations, as estimated from the molar mass determined by GPC analysis. The solution was infused into the ESI source at a rate of 5 μl min−1 with a Harvard syringe pump. Typical ESI conditions were: capillary voltage: 3.1 kV; cone voltage: 80 V; source temperature: 80 °C and desolvation temperature: 120 °C. Dry nitrogen was used as the ESI gas. The quadrupole was set to pass ions from 100 to 3000 Th and all ions were transmitted into the pusher region of the time-of-flight analyzer where they were mass-analyzed with a 1 s integration time. Data were acquired in continuum mode until acceptable averaged data were obtained. Size exclusion chromatography (SEC/GPC) was performed in THF at 35 °C using a Polymer Laboratories liquid chromatograph equipped with a PL-DG802 degasser, an isocratic HPLCpumpLC 1120 (flow rate = 1ml min−1), a triple detector: refractive index (ERMA 7517), capillary viscometry and light scattering RALS (Viscotek T-60) (Polymer Laboratories GPC-RI/CV/RALS), an automatic injector (Polymer Laboratories GPC-RI/UV) and four columns: a PL gel 10 μm guard column and three PL gel Mixed-B 10μm columns. PS standards were used for calibration.General polymerization procedure
All polymerizations were performed in an inert atmosphere glovebox using previously dried vials. As an illustrative example: LA (200 mg, 1.4 mmol), and ICl3 (30 mg, 0.13 mmol) were dissolved in CHCl3 (0.3 g). The mixture was allowed to dissolved for 30 s in a screwed capped vial before the addition of 11-BU (2 mg, 0.008 mmol). The polymerization was stopped after 44 h by addition of few drops of acetone, precipitated in cold methanol, analyzed by 1H NMR and SEC.Acknowledgements
This work was partially supported by the Belgian Federal Science Policy Office (PAI6/27). O.C. is a Research Associate of the Belgian National Fund for Scientific Research (FRS-FNRS). Dr P. Gerbaux is gratefully thanked for the ESI-MS measurements.Notes and references
- (a) P. Metrangolo and G. Resnati, Science, 2008, 321, 918 CrossRef CAS; (b) P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Angew. Chem., Int. Ed., 2008, 47, 6114 CrossRef CAS.
- (a) A. Casnati, R. Liantonio, P. Metrangolo, G. Resnati, R. Ungaro and F. Ugozzoli, Angew. Chem., Int. Ed., 2006, 45, 1915 CrossRef CAS; (b) R. Liantonio, P. Metrangolo, F. Meyer, T. Pilati, W. Navarrini and G. Resnati, Chem. Commun., 2006, 1819 RSC; (c) P. Metrangolo, F. Meyer, T. Pilati, D. M. Proserpio and G. Resnati, Chem.–Eur. J., 2007, 13, 5765 CrossRef.
- (a) T. Caronna, R. Liantonio, T. A. Logothetis, P. Metrangolo, T. Pilati and G. Resnati, J. Am. Chem. Soc., 2004, 126, 4500 CrossRef CAS; (b) H. L. Nguyen, P. N. Horton, M. B. Hursthouse, A. C. Legon and D. W. Bruce, J. Am. Chem. Soc., 2004, 126, 16 CrossRef CAS; (c) T. Takeuchi, Y. Minato, M. Takase and H. Shinmori, Tetrahedron Lett., 2005, 46, 9025–9027 CrossRef CAS; (d) E. Cariati, A. Forni, P. Metrangolo, F. Meyer, G. Resnati, S. Righetto, E. Tordin and R. Ugo, Chem. Commun., 2007, 2590 RSC; (e) Y. Lu, T. Shi, Y. Wang, H. Yang, X. Yan, X. Luo, H. Jiang and W. Zhu, J. Med. Chem., 2009, 52, 2854 CrossRef CAS.
- F. Cataldo, Eur. Polym. J., 1996, 32, 1297 CrossRef CAS and references therein.
- (a) F. C. Pigge, V. R. Vangala, P. P. Kapadia, D. C. Swenson and N. P. Rath, Chem. Commun., 2008, 4726 RSC; (b) P. Auffinger, F. A. Hays, E. Westhof and P. S. Ho, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16789 CrossRef CAS; (c) A. R. Voth, F. A. Hays and P. S. Ho, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6188 CrossRef CAS.
- (a) K. Maruoka, Pure Appl. Chem., 2002, 74(1), 123 CrossRef CAS and references therein; (b) M. Labet and W. Thielemens, Chem. Soc. Rev., 2009, 38, 3484–3504 RSC; (c) Ch.M. Thomas, Chem. Soc. Rev., 2010, 39, 165 RSC; (d) A. Arbaoui and C. Redshaw, Polym. Chem., 2010 10.1039/b9py00334g.
- (a) A. P. Dove, R. C. Pratt, B. G. G. Lohmeijer, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2005, 127, 13798–13799 CrossRef CAS; (b) O. Coulembier, D. P. Sanders, A. Nelson, A. N. Hollenbeck, A. W. Horn, J. E. Rice, M. Fujiwara, Ph. Dubois and J. L. Hedrick, Angew. Chem., 2009, 121, 5272–5275 CrossRef.
- The concomittant presence of a minimum frequency band relative to the νCO of the lactide free of interaction is always present at ∼1756 cm−1 for a [ICl3]0/[LA]0 < 2.
- It is still not clear if more than one ICl3 is required for the O-acyl cleavage of the LA monomer.
| This journal is © The Royal Society of Chemistry 2010 |