Definition of the uptake mechanism and sub-cellular localisation profile of emissive lanthanide complexes as cellular optical probes†
Elizabeth J.
New
,
Aileen
Congreve
and
David
Parker
*
Department of Chemistry, Durham University, South Road, Durham, UK DH1 3LE. E-mail: david.parker@dur.ac.uk
First published on 12th May 2010
Abstract
A series of experiments has been undertaken in order to gain a greater understanding of the cellular uptake and localisation behaviour of emissive lanthanide complexes as cellular stains or probes. Out of a large number of structurally related complexes characterised recently, a set of seven representative examples has been examined in detail, containing either tetraazatriphenylene or azaxanthone-based sensitising chromophores. Intracellular localisation profiles and cellular uptake and egress behaviour have been studied by microscopy and flow cytometry. Typically, the maximum intracellular concentration was of the order of 0.4 mM, or about 109 complexes per cell. The complexes studied were generally not toxic and did not perturb the mitochondrial membrane potential. A common uptake mechanism of macropinocytosis has been identified. A generalisation of trends in behaviour, and structure–activity relationships is presented, and the implications for future probe design discussed.
Introduction
A major aspect of molecular imaging concerns the in vivo characterisation and measurement of processes at the cellular and molecular level, allowing the study of biological processes in living cells.1 Amongst molecular imaging techniques, optical imaging allows very high spatial resolution and good sensitivity.2 The design of responsive cellular probes for use in optical imaging remains a key challenge for biology and medicine. Existing classes of probes in widespread use, such as fluorescent organic dyes,3 fluorescent proteins4 and quantum dots,5 have showed great promise, but their utility is limited by their small Stokes' shifts, short luminescence lifetimes and broad and formless emission profiles.6 Luminescent europium or terbium complexes have proved to be viable alternatives,7 with their information-rich emission profiles and long luminescence lifetimes allowing the ‘gating out’ of interfering background signals.7a The incorporation of a sensitising chromophore into the ligand ensures a high molar absorptivity and large Stokes' shift.8A common theme of the study of luminescent lanthanide complexes has been the design of systems which can report on their environment. To this end, systems have been reported which are sensitive to pH,9 endogenous anions such as bicarbonate,10 citrate11 and urate,12 and metal ions such as zinc,13 copper(I)14 and potassium.15 In addition, lanthanide complexes have been developed as potential probes for sub-cellular events, including redox state,16 peptide phosphorylation17 and singlet oxygen.18 Most valuable amongst such complexes are systems which can act as ratiometric probes, and can therefore provide information about the target analyte independent of probe concentration and the local environment. In general, studies of these complexes have been performed in vitro, and have not demonstrated in cellulo applicability.
In order for a sensitised lanthanide complex to have use as a cellular probe, it must exhibit certain biological properties. Notably, the complex must readily cross the cell membrane, and localise in a region of interest within the cell. For example, a pH sensor might be best localised in the lysosomes, in which acidity can signify endosome age or health.19 Similarly, a probe for phosphorylation might be best directed to the mitochondria, where numerous phosphorylation events take place. In addition, a complex that is intended to act as a probe of cellular function should only minimally perturb the cellular homeostasis, and in particular should not induce cell proliferation or lead to death at working concentrations.
The importance of these biological properties has led to recent efforts to characterise the sub-cellular behaviour of a large number of sensitised lanthanide complexes.20 This led to the identification of a number of important structural effects; the variation of sub-cellular localisation with the nature of the sensitising chromophore;21 the conservation of localisation following changes to either the ligand pendant arms, complex charge or helicity;22a the alteration of protein binding and quenching sensitivity that occur following minor structural changes.22 In addition, most complexes were reported to be non-toxic at dosing concentrations, with only isolated cytotoxic complexes.21a,23 Every complex investigated exhibited noticeable cell uptake, and preliminary studies of a set of sensitised lanthanide complexes were consistent with an uptake mechanism of macropinocytosis.24
Here, we describe experiments performed in order to gain a greater understanding of the cellular behaviour of luminescent lanthanide complexes. Seven representative ligand sets are examined, containing tetraazatriphenylene and azaxanthone-based sensitising chromophores. A generalisation of trends in behaviour, and structure–activity relationships is presented, and the implications for future probe design discussed.
Results and discussion
The cellular localisation profile of complexes was examined in CHO or NIH 3T3 cells, with complex concentrations of 10 to 100 μM, for incubation times of between 5 min and 24 h. The distribution of the complex was observed by examining the lanthanide luminescence by fluorescence microscopy, following excitation of the chromophore. Optical sections throughout the cell confirmed that the complex was localised within the cell, and not concentrated at the cell membrane. Sub-cellular localisation was confirmed by the use of commercially available cellular stains (Invitrogen) for the lysosomes, mitochondria, nucleus and nucleolus. All complexes studied could be classified into four groups according to their sub-cellular localisation (Fig. 1). While the complexes in each group were found to have remarkably similar biological properties, each class exhibited distinct behaviour. The four classes and their principal characteristics will be discussed briefly.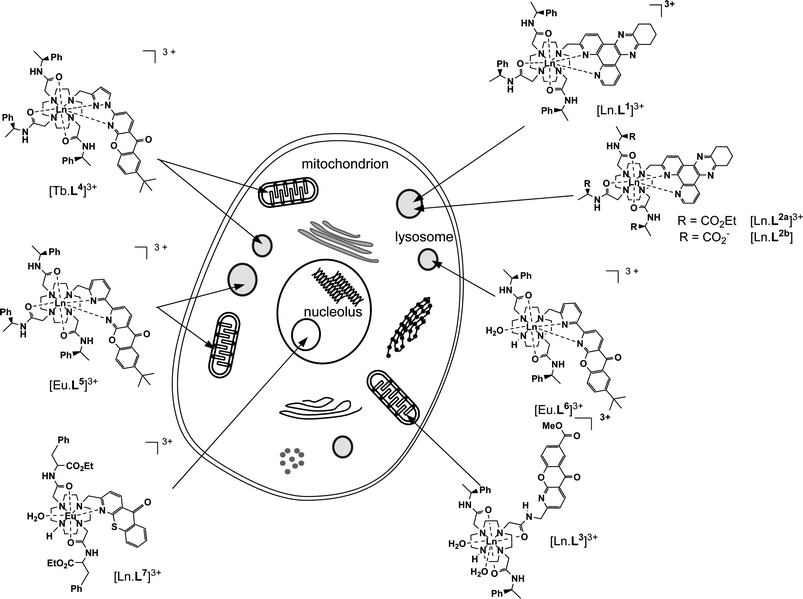 | ||
| Fig. 1 The four distinct sub-cellular localisation profiles of luminescent lanthanide complexes—lysosomal, mitochondrial, nucleolar and mitochondrial-lysosomal—with representative examples of complexes exhibiting each behaviour. | ||
Classes of localisation behaviour
The largest group of complexes, including [Ln.L1]3+, [Ln.L2a]3+, [Ln.L2] and [Eu.L6]3+, are those which are observed to localise to the lysosomes. This sub-cellular distribution was confirmed by co-staining with LysoTracker probes (Invitrogen; Fig. 2). The lysosomal localisation was observed at all incubation times, from 5 min to 24 h. A smaller group of complexes, including [Ln.L3]3+, has previously been reported to exhibit mitochondrial localisation.21a This distribution was observed for incubation times from 5 min to 12 h, and was confirmed by staining with MitoTracker probes. At incubation times longer than 24 h, the luminescence pattern was no longer consistent with a mitochondrial distribution, but instead co-localised with LysoTracker probes. It appears, therefore, that after longer periods of time, the complexes are being transported to the lysosomes for degradation and removal from the cell.![Fluorescence microscopy images of CHO cells treated with various europium complexes (50 μM, 4 h) and commercially-available fluorescent stains (30 min) showing the lysosomal localisation of [Eu.L1]Cl3, the mitochondrial localisation of [Eu.L3]Cl3, the mitochondrial-lysosomal co-localisation of [Eu.L5]Cl3 and the nucleolar distribution of [Eu.L7]Cl3. (A) Europium luminescence, (B) stain fluorescence and (C) image overlay. Scale bars represent 20 μm; image brightness and contrast have been adjusted.](/image/article/2010/SC/c0sc00105h/c0sc00105h-f2.gif) | ||
| Fig. 2 Fluorescence microscopy images of CHO cells treated with various europium complexes (50 μM, 4 h) and commercially-available fluorescent stains (30 min) showing the lysosomal localisation of [Eu.L1]Cl3, the mitochondrial localisation of [Eu.L3]Cl3, the mitochondrial-lysosomal co-localisation of [Eu.L5]Cl3 and the nucleolar distribution of [Eu.L7]Cl3. (A) Europium luminescence, (B) stain fluorescence and (C) image overlay. Scale bars represent 20 μm; image brightness and contrast have been adjusted. | ||
In contrast to these two, well-defined classes of complexes, a third distinct group of complexes has been identified, in which luminescence is observed simultaneously in both the lysosomes and the mitochondria. In such cases, bright spots consistent with lysosomal staining can be observed, as well as the more diffuse pattern characteristic of mitochondrial localisation. The sub-cellular distribution was confirmed by co-staining with both LysoTracker and MitoTracker probes (Fig. 2). Such behaviour is demonstrated by complexes [Tb.L4]3+ and [Eu.L5]3+.
The final class of complexes is those which localised to the nucleoli, the sub-nuclear, protein-rich structure which is primarily involved in ribosome assembly.25 This distribution was observed at normal dosing concentrations for a number of complexes, including [Ln.L7]3+. In addition, nucleolar distribution at high incubation concentrations was observed for a number of complexes which, at lower concentrations, localised to the lysosomes.26 Previous work has demonstrated that this nucleolar localisation can be attributed to an enhanced cellular membrane permeability, induced by the presence of the probe itself.24
Structural factors affecting localisation
In order to rationally design sensitised lanthanide complexes with a specific sub-cellular localisation, it is important to establish any relationships between the structure of a complex and its localisation profile. There are a number of structural aspects which could affect the intracellular distribution of the complex: the sensitising chromophore itself, the additional carboxylate, amide or phosphinate pendant arms, and the lanthanide ion itself. In addition, the geometry, charge and counterion of the complex could influence the sub-cellular distribution. In order to assess the importance of each structural component, a series of complexes were studied in which only one feature was varied. Aspects of such investigations have been reported in part,21a,22 and are summarised here.There are a number of ways in which the chromophore can be altered in the synthesis of luminescent lanthanide complexes. The nature of the chromophore appears to have an effect on the localisation of the complex. This is evident in the varying distributions of [Tb.L1]3+ and [Tb.L4]3+ which differ only in the sensitising chromophore. Small modifications to the chromophore structure, on the other hand, do not appear to influence the complex localisation.21a,22b A marked change in localisation has been observed on alteration of the linkage between the chromophore and the cyclen ring. This has previously been reported for the mitochondrially-localising [Ln.L3]3+ and its lysosomally-localising analogue in which the amide linkage is replaced by a methylene group.21a This is also evident in the case of the analogue [Eu.L6]3+, containing a pyridine linkage, which localises to the lysosomes.
In considering the effect of the pendant arms on the localisation of complexes, it is important to consider both the nature and number of pendant arms. Complexes [Ln.L1]3+ and [Ln.L2a]3+, which form part of a larger group of complexes previously discussed,22a differ only in the identity of the amide-based pendant arms, and exhibit identical lysosomal localisation. In contrast, the number of pendant arms, or the ligand denticity, does appear to influence the cellular distribution of the complex. While [Eu.L5]3+ is observed in both lysosomes and mitochondria, removal of one pendant arm gives rise to [Eu.L6]3+, which exhibits a purely lysosomal distribution. This behaviour has also been observed for the analogue of [Tb.L4]3+ bearing only two pendant arms.
In every case, Tb and Eu complexes of a common ligand exhibited identical sub-cellular localisation patterns, indicating that the nature of the lanthanide ion does not affect distribution. There is no evidence to suggest that complex charge has an effect on sub-cellular localisation: [Ln.L2a]3+ and its neutral analogue [Ln.L2b], for example, exhibit lysosomal distributions. Previous studies of complexes with differing geometries and helicities have not shown any effect on localisation profiles.22a The counterion which was most commonly employed for cationic complexes is chloride. Cellular studies were also performed in which acetate, nitrate and triflate forms of the complexes were administered to cells. These did not reveal any significant change in the sub-cellular localisation.
In summary, these investigations have revealed that the localisation of a complex is affected by the nature of the chromophore and, more significantly, its point of attachment to the macrocycle, as well as by the ligand denticity. It is therefore important to consider whether it is possible to predict the sub-cellular localisation of a novel complex. The complexes which have been investigated comprise a library of chromophores and pendant arms, which is not exhaustive. The information which has been gained from this structural study allows the prediction of the localisation of a complex composed of elements from this library. For entirely new complexes, however, which contain different chromophores or pendant arms, such prediction is less straightforward. In addition, if alternative sub-cellular localisations were desired, it is likely that more drastic structural changes would need to be made, or a targeting moiety incorporated into the complex. While the nature of the pendant arms does not appear to have any effect on the sub-cellular distribution, complexes bearing three identical pendant arms, such as [Tb.L4]3+ and [Eu.L5]3+, appear to exhibit less well-defined localisation profiles than their analogues in which one pendant arm has been removed, leaving a free cyclen NH.
Cellular uptake profile
An indication of the kinetics of cellular uptake was obtained by measuring the lanthanide concentration within cells after various incubation times, and found to be similar for all complexes. The behaviour of a typical complex, [Tb.L1]Cl3, is shown in Fig. 3. The intracellular lanthanide concentration was determined by ICP-MS, and calculated with reference to the protein concentration, determined by the BCA assay (Sigma Aldrich). While this assay does not allow for calculation of an absolute intracellular concentration, the protein content is proportional to the number of cells, and allows for comparison of samples. This method was found to circumvent the loss of complex that was found to accompany other methods of analysis, such as flow cytometric counting and sorting.![Uptake and egress of [Tb.L1]Cl3 (50 μM) in NIH 3T3 cells over time, showing standard deviations. (a) Shows uptake of complex over a 24 h period; (b) shows the egress of complex following 4 h incubation over a 4 h period.](/image/article/2010/SC/c0sc00105h/c0sc00105h-f3.gif) | ||
| Fig. 3 Uptake and egress of [Tb.L1]Cl3 (50 μM) in NIH 3T3 cells over time, showing standard deviations. (a) Shows uptake of complex over a 24 h period; (b) shows the egress of complex following 4 h incubation over a 4 h period. | ||
In control experiments examining the protein concentration of sets of counted cells, it was estimated that one hundred micrograms of protein corresponded to 250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells. Therefore if we consider an intracellular lanthanide ion concentration of 1 nmol mg−1 protein, this corresponds to 4 × 10−16 moles/cell, or 2.4 × 108 lanthanide complexes per cell. Assuming an average cell volume of 5000 cubic microns (5 × 10−9 cc), then a concentration of complex within the cell of 1 nmol mg−1 protein is of the order of 80 μM. Similar values have been estimated previously.26
000 cells. Therefore if we consider an intracellular lanthanide ion concentration of 1 nmol mg−1 protein, this corresponds to 4 × 10−16 moles/cell, or 2.4 × 108 lanthanide complexes per cell. Assuming an average cell volume of 5000 cubic microns (5 × 10−9 cc), then a concentration of complex within the cell of 1 nmol mg−1 protein is of the order of 80 μM. Similar values have been estimated previously.26
The uptake profile (Fig. 3a) indicates that the intracellular concentration increases very rapidly over the first four hours, with only a slight increase after this time-point. An incubation time of 4 h was therefore selected for all subsequent uptake experiments. The egress (Fig. 3b) profile was established by incubating the cells with 50 μM complex for 4 h, after which the medium was removed and replaced by buffered saline solution. Egress was relatively rapid over the first 20 min, but by one hour appeared to reach an equilibrium value and had decreased very little at 4 h. It may be noted that the maximum intracellular concentration defined here is of the order of 0.4 mM. This lies comfortably within the target range for labelling cells with Gd complexes, for cell tracking MRI applications.
Mechanisms of cellular uptake
The mode of cellular uptake was previously reported for three complexes in a preliminary communication, and was found to be consistent with the mechanism of macropinocytosis.36 In order to confirm this finding, the experiment was repeated on a larger set of complexes which represented different sub-cellular behaviours: the lysosomally-localising [Eu.L1]3+ and [Tb.L2a]3+; mitochondrially-distributed [Eu.L3]3+; [Tb.L4]3+ and [Eu.L5]3+ which localise in both lysosomes and mitochondria, and [Eu.L7]3+ which exhibits a nucleolar distribution. CHO cells were treated with 50 μM complex for 4 h, washed and harvested, and the intracellular lanthanide concentration determined by ICP-MS and the BCA assay. In order to assess the contribution of various endocytotic pathways, inhibitors or activators of these processes were selected (Fig. 4): sucrose (inhibition of clathrin-mediated endocytosis),27 chlorpromazine (inhibitory interaction with clathrin),28 filipin (perturbation caveolae by sequestering cholesterol),29 wortmannin (inhibition of macropinocytosis through blocked PI-3 kinase),30 amiloride (inhibition of macropinocytosis by blocked Na+/H+ pump),31 fatty acid glycerols (stimulation of macropinocytosis by activating protein kinase C),32 phorbol esters (stimulation of macropinocytosis),32b monensin (prevention of endosomal acidification and maturation)33 and chloroquine (increase of endosomal pH).34 In addition, two non-specific inhibitors were also utilised: poly-L-lysine (disruption of cell-membrane association)35 and low temperature (blocking energy dependent processes).36 Cells were exposed to these treatments for 30 min prior to addition of the lanthanide complex.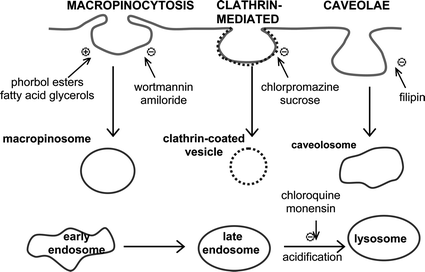 | ||
| Fig. 4 Schematic diagram illustrating the three endocytotic pathways in addition to the intracellular maturation of endosomes to lysosomes. Inhibitors (−) and activators (+) of each pathway are indicated. | ||
The measured intracellular concentrations, relative to the control cells treated with complex alone, demonstrate behaviour that is consistent with uptake by macropinocytosis. Both wortmannin and amiloride resulted in statistically significant decreased uptake of all complexes (p < 0.01 and < 0.05 are indicated, Fig. 5), while the presence of the phorbol ester and fatty acid glycerol demonstrably enhanced uptake. The other drug treatments did not result in significant variations from the control. This finding is consistent with the observation that at short incubation times (5–20 min) these luminescent lanthanide complexes co-localise with FITC–dextran, a fluorescent marker of the macropinosomes.24
![Relative changes in uptake of [Eu.L1]Cl3 (blue), [Tb.L2b] (red), [Eu.L3]Cl3 (green), [Tb.L4]Cl3 (yellow), [Eu.L5]Cl3 (purple) and [Eu.L7]Cl3 (pink) in the presence of various inhibitors and activators. Statistically significant differences from controls (* p < 0.05, ** p < 0.01) calculated using a two-tailed, paired Student's t-test.](/image/article/2010/SC/c0sc00105h/c0sc00105h-f5.gif) | ||
| Fig. 5 Relative changes in uptake of [Eu.L1]Cl3 (blue), [Tb.L2b] (red), [Eu.L3]Cl3 (green), [Tb.L4]Cl3 (yellow), [Eu.L5]Cl3 (purple) and [Eu.L7]Cl3 (pink) in the presence of various inhibitors and activators. Statistically significant differences from controls (* p < 0.05, ** p < 0.01) calculated using a two-tailed, paired Student's t-test. | ||
Confirmation of an active uptake mechanism
The observation that cellular uptake appears to occur via a mechanism of macropinocytosis suggests that transport across the cell membrane follows an active mechanism. This is confirmed by the marked decrease in lanthanide uptake in cells incubated at 4 °C (Fig. 5). Further confirmation of an active uptake mechanism can be gained from a study of the uptake of [Tb.L4]Cl3 and [Eu.L5]Cl3 at various concentrations following 4 h incubation (Fig. 6). In each case, at low dosing concentrations, the intracellular lanthanide concentrations increase with increasing incubation concentrations before reaching a plateau. This is consistent with a saturation of the available sites for macropinocytosis on the cell surface. At very high dosing concentrations, well above those employed for microscopy studies, and above the IC50 values of the two complexes (Table 1), significantly increased uptake is observed. This could indicate that at these high concentrations, there is some passive diffusion of the complex across the cell membrane. If, on the other hand, uptake was occurring by a passive diffusion mechanism, uptake would be expected to increase linearly with concentration for all dosing concentrations.![Intracellular lanthanide concentrations as a function of dosing concentrations for CHO cells incubated for 4 h with (a) [Tb.L4]Cl3 and (b) [Eu.L5]Cl3.](/image/article/2010/SC/c0sc00105h/c0sc00105h-f6.gif) | ||
| Fig. 6 Intracellular lanthanide concentrations as a function of dosing concentrations for CHO cells incubated for 4 h with (a) [Tb.L4]Cl3 and (b) [Eu.L5]Cl3. | ||
| Log Poct | SD | IC50/μM | SD | |
|---|---|---|---|---|
| a n.d. = not determined; the IC50 values obtained using the parallel WST-1 method (see ESI) for [Eu.L3]Cl3, [Tb.L4]Cl3 and [Eu.L7](OTf)3 were >200, 40 [± 6] and 59 [± 10] μM respectively, comparable to the values obtained by the MTT assay. | ||||
| [Eu.L1]Cl3 | −1.34 | 0.10 | 109 | 20 |
| [Tb.L1]Cl3 | n.d. | — | 131 | 27 |
| [Eu.L2a]Cl3 | −2.10 | 0.40 | 144 | 12 |
| [TbL2a]Cl3 | −2.06 | 0.09 | n.d. | — |
| [Eu.L2b] | −1.74 | 0.37 | >240 | — |
| [TbL2b] | −1.56 | 0.25 | n.d. | — |
| [Eu.L3]Cl3 | −1.56 | 0.09 | 164 | 31 |
| [Tb.L4]Cl3 | 0.05 | 0.04 | 40.9 | 8.9 |
| [Eu.L5]Cl3 | −0.06 | 0.03 | 72.9 | 4.1 |
| [Eu.L7](OTf)3 | 1.06 | 0.13 | 79.0 | 9.4 |
| [Eu.L7]Cl3 | 0.49 | 0.11 | 120 | 10 |
The possibility of a passive uptake mechanism may also be addressed by examining the lipophilicities of a range of complexes. Values of log P were obtained by allowing the complex to partition between 1-octanol and water, and calculating the concentration in each layer by measuring the lanthanide emission relative to a calibration curve. The measured partition coefficients range from hydrophilic (−2.10) to lipophilic (+1.06). The nature of the lanthanide ion does not appear to affect the log P value, but variation of the counterion does have a significant effect, with the triflate form of [Eu.L7]3+ being more lipophilic than the chloride form. This is consistent with the observed lower aqueous solubility of triflate salts.
While the complexes vary greatly in partitioning behaviour, microscopy studies revealed a similar rate of uptake for each complex, with luminescence readily observed 5 min after dosing. This confirms that cell uptake is most unlikely to occur by passive diffusion as in such a case diffusion rates would be likely to mirror trends in log P.
Cellular toxicity of complexes
In order for a complex to act as a cellular probe, it is essential that it is non-toxic at the concentrations at which it will be used. It is therefore important to assess the toxicity of all potential probes, and to direct studies towards non-toxic candidates. The cytotoxicity of the sensitised lanthanide complexes towards NIH 3T3 cells after 24 h incubation was assessed by the spectrophotometric MTT assay based on perturbation of mitochondrial redox. For each complex, an IC50 value was calculated as the concentration which inhibits viability by 50% compared to the untreated, control cells.Analysis of the resulting IC50 values (Table 1) highlights a number of trends. The replacement of Eu3+ for Tb3+ does not greatly alter toxicity, exemplified by the study of two complexes of L1. In general, it appears that neutral complexes are less cytotoxic than their cationic analogues; [Eu.L2a]Cl3, for example, exhibits an IC50 value of 144 μM, while its neutral analogue, [Eu.L2b] was non-toxic over the range studied. The nature of the sensitising chromophore also affects the cytotoxicity; while the tricationic complexes, [Eu.L1]Cl3 and [Eu.L2a]Cl3, containing a tetraazatriphenylene chromophore exhibit IC50 values of greater than 100 μM, [Eu.L5]Cl3, which contains a pyridine-linked azaxanthone sensitiser, is more toxic. Notably, it was observed that complexes containing an appended tBu group, such as [Tb.L4]Cl3 and [Eu.L5]Cl3, exhibit higher cytotoxicity. The difference in cytotoxicity between the triflate and chloride salts of [Eu.L7]3+ also indicates that the nature of the counterion may play an important role. This result, coupled to the lower solubility of the triflate salts, indicates that triflates are not the preferred salts for administration of complexes.
The cytotoxicity results indicate that complexes are generally non-toxic under the conditions which are most applicable to their use as cellular probes; incubation times of 1 to 4 h, and dosing concentrations of up to 50 μM. Previous studies have reported isolated examples of complexes which exhibited higher toxicities.21a, 23 In each case, this high cytotoxicity could be explained by demonstrable mechanisms, such as dissociation of the complex to toxic metabolites and disruption of cell integrity by insertion into the cell membrane. Such cases, however, appear to be the exception rather than the rule and can be easily avoided.
Mitochondrial membrane potential
The most cytotoxic complexes of those presented in Table 1 are [Tb.L4]Cl3 and [Eu.L5]Cl3. These complexes exhibit similar sub-cellular distribution, localising in both the lysosomes and mitochondria. In order to further study their cellular effects, the mitochondrial membrane potential (MMP) of cells treated with these complexes was assessed by a flow cytometry assay which measures the green fluorescence of MitoTracker Green (MTG; Invitrogen) against the red fluorescence of chloromethyl-X-rosamine (CMXRos; Invitrogen).37 While MTG stains every mitochondrion, independent of its MMP, the mitochondrial uptake of CMXRos is dependent on the MMP. CHO cells were treated with 50 μM complex for varying incubation times and MTG and CMXRos added for the final 30 min (200 nM). Cells were then harvested and analysed by flow cytometry, measuring green (FL1) against red (FL4) fluorescence. In addition to [Tb.L4]Cl3 and [Eu.L5]Cl3, the effect of the mitochondrially-localising, non-toxic [Eu.L3]Cl3 was also explored. Carbonyl cyanide p-trifluoromethoxylphenylhydrazone (FCCP), an uncoupling agent which readily destroys the MMP,38 was employed as a positive control. As expected, treatment with increasing concentrations of FCCP led to disruption of the MMP, and hence a lower FL4/FL1 ratio compared to the untreated control cells (Fig. 7a). Cells treated with [Eu.L3]Cl3 do not exhibit a decreased red fluorescence, suggesting that the complex does not disrupt the MMP. This complex therefore appears to localise in the mitochondria without disrupting its membrane potential, confirming the potential for its use as a probe of normal mitochondrial conditions. The remaining two complexes, on the other hand, elicit more noticeable changes in MMP, giving rise to FL4/FL1 intensities which are less than for untreated cells at all time points. For [Tb.L4]Cl3, this ratio decreases with time, while for [Eu.L5]Cl3, the FL4/FL1 ratio indicates a greater uncoupling at 4 h, with partial restoration of the MMP after 24 h.![(a) FL4/FL1 mean intensity from flow cytometric analysis of CHO cells with various treatments. Carbonyl cyanide p-trifluoromethoxylphenylhydrazone (FCCP) treatments are 20 μM (black) and 100 μM (grey) for 3 h; [Eu.L3]Cl3 treatment is 50 μM for 1 h (black) and 4 h (grey); [Tb.L4]Cl3 and [Eu.L5]Cl3 treatments are 50 μM for 4 h (black) and 24 h (grey). (b) and (c) flow cytometry ‘dot’ plots following staining with propidium iodide and annexin V–FITC of CHO cells treated for 24 h with (b) 200 μM [Tb.L4]Cl3 and (c) 200 μM [Eu.L5]Cl3.](/image/article/2010/SC/c0sc00105h/c0sc00105h-f7.gif) | ||
| Fig. 7 (a) FL4/FL1 mean intensity from flow cytometric analysis of CHO cells with various treatments. Carbonyl cyanide p-trifluoromethoxylphenylhydrazone (FCCP) treatments are 20 μM (black) and 100 μM (grey) for 3 h; [Eu.L3]Cl3 treatment is 50 μM for 1 h (black) and 4 h (grey); [Tb.L4]Cl3 and [Eu.L5]Cl3 treatments are 50 μM for 4 h (black) and 24 h (grey). (b) and (c) flow cytometry ‘dot’ plots following staining with propidium iodide and annexin V–FITC of CHO cells treated for 24 h with (b) 200 μM [Tb.L4]Cl3 and (c) 200 μM [Eu.L5]Cl3. | ||
In order to further understand these effects, an apoptosis–necrosis assay was performed, in which propidium iodide (PI) was employed as a marker for necrosis, and an annexin V–FITC conjugate used to signal apoptosis.39 CHO cells were treated with 200 μM complex for 24 h, above the IC50 value of each complex. The cells were then harvested, treated with PI and annexin V–FITC, and analysed by flow cytometry, measuring red (FL4; PI) against green (FL1; FITC) fluorescence (Fig. 7b and c). The flow cytometry plots indicate that a population of cells treated with [Tb.L4]Cl3 are stained with PI but do not exhibit binding to annexin V, indicative of necrosis. In contrast, cells treated with [Eu.L5]Cl3 show both red and green fluorescence, suggesting cell death by apoptosis. These modes of cell death are consistent with the observed mitochondrial membrane changes. Mitochondrial membrane damage is an effect rather than a cause of necrosis, and as a result, the MMP should decrease over time, as observed for [Tb.L4]Cl3. Apoptosis, on the other hand, is driven by disruptions to the mitochondria, and a more rapid change in MMP for [Eu.L5]Cl3 is therefore observed. Many of these cells with dramatically perturbed MMP at 4 h would have completed apoptosis by 24 h and would be too fragmented to be measured by flow cytometry, resulting in a higher average MMP. This investigation provide further insight into the low IC50 values of [Tb.L4]Cl3 and [Eu.L5]Cl3, suggesting that their toxicity is related to a perturbation of the mitochondrial membrane potential. The results add further weight to the argument that complexes which localise in both mitochondria and lysosomes are not ideal candidates for use as cellular probes.
Summary and conclusions
The design of compounds or complexes for use as sub-cellular probes or drugs often employs strategies to induce certain cellular behaviour. For example, compounds might be encapsulated in liposomes to enhance cellular uptake, or conjugated to cell penetrating peptides to improve uptake and localisation. However, these strategies may also alter properties such as selectivity, toxicity or efficacy. It is therefore important to assess whether such modifications are required, or whether cellular behaviour of the native system is sufficient to achieve its purpose. For the sensitised lanthanide complexes studied, it is evident that no such strategy need be employed to induce cellular uptake. The range of complexes studied was observed to be transported readily across the cell membrane. It is likely that any complex of a similar structure (bearing a cyclen macrocycle and a polycyclic heterocyclic sensitiser) will undergo macropinocytosis. Future probe design therefore need not concentrate on inducing cellular uptake. Similarly, the majority of complexes are not cytotoxic towards the cell lines tested, and it is not necessary to direct concerted efforts towards the design and synthesis of non-toxic probes.Sensitised lanthanide complexes appear to exhibit one of a limited number of sub-cellular distribution patterns, principally localising in the lysosomes or mitochondria. In addition, nucleolar localisation can be promoted in cases of enhanced membrane permeability. While these membrane effects may perturb the homeostasis of the cell, this technique may prove to be a valuable method by which to probe this protein-rich region of the cell.
The results presented here indicate that if lysosomal or mitochondrial localisation is required, it is not necessary to tag complexes to direct their delivery. Cells can be labelled with lanthanide complexes in concentrations of the order of 0.05 to 0.5 mM. Such levels are not only sufficient for optical signalling using one- or two-photon excitation, but also are in the target range for the cell loading levels required for tracking cell populations by MRI, assisted by Gd contrast agents.
Clear structural requirements for mitochondrial localisation have also been defined, while a lysosomal localisation appears to be the default distribution. In addition, structural features have been identified which evade the localisation of the complex in both mitochondria and lysosomes. If other localisations are desired, however, it may be necessary to investigate more drastic structural modifications, or to conjugate the complex to a moiety with a controlling influence on uptake and sub-cellular trafficking.
Acknowledgements
We thank the Association of Commonwealth Universities for a scholarship.References
- R. Weissleder and U. Mahmood, Radiology, 2001, 219, 316–333 Search PubMed; T. F. Massoud and S. S. Gambhir, Genes Dev., 2003, 17, 545–580 CrossRef CAS.
- R. Heintzmann and G. Ficz, Briefings Funct. Genomics Proteomics, 2006, 5, 289–301 Search PubMed.
- L. D. Lavis and R. T. Raines, ACS Chem. Biol., 2008, 3, 142–155 CrossRef CAS.
- R. Y. Tsien, Annu. Rev. Biochem., 1998, 67, 509–544 CrossRef CAS.
- W. C. W. Chan, D. J. Maxwell, X. Gao, R. E. Bailey, M. Han and S. Nie, Curr. Opin. Biotechnol., 2002, 13, 40–46 CrossRef CAS.
- R. P. Haugland, A Guide to Fluorescent Probes and Labelling Technologies, Molecular Probes, Eugene, Oregon, 10th edn, 2005 Search PubMed; P. Abbyad, W. Childs, X. Shi and S. G. Boxer, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20189–20194 Search PubMed.
- (a) D. Parker and J. A. G. Williams, in Metal Ions in Biological Systems – The Lanthanides and Their Interrelations with Biosystems, ed. A. Sigel and H. Sigel, Marcel Dekker, New York, 2003 Search PubMed; (b) S. Faulkner, S. J. A. Pope and B. P. Burton-Pye, Appl. Spectrosc. Rev., 2005, 40, 1–31 CrossRef CAS; (c) S. Pandya, J. Yu and D. Parker, Dalton Trans., 2006, 2757–2766 RSC.
- J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC.
- D. Parker, K. Senanayake and J. A. G. Williams, Chem. Commun., 1997, 1777–1778 RSC; D. Parker, K. Senanayake and J. A. G. Williams, J. Chem. Soc., Perkin Trans. 2, 1998, 2129–2139 RSC; A. P. de Silva, H. Q. N. Gunaratne and T. E. Rice, Angew. Chem., Int. Ed. Engl., 1996, 35, 2116–2118 CrossRef CAS; R. Pal and D. Parker, Chem. Commun., 2007, 474–476 RSC.
- Y. Bretonniere, M. J. Cann, D. Parker and R. Slater, Chem. Commun., 2002, 1930–1931 RSC; Y. Bretonniere, M. J. Cann, D. Parker and R. Slater, Org. Biomol. Chem., 2004, 2, 1624–1632 RSC.
- D. Parker and J. Yu, Chem. Commun., 2005, 3141–3143 RSC.
- R. A. Poole, F. Kielar, S. L. Richardson, P. A. Stenson and D. Parker, Chem. Commun., 2006, 4084–4086 RSC.
- O. Reany, T. Gunnlaugsson and D. Parker, Chem. Commun., 2000, 473–474 RSC; O. Reany, T. Gunnlaugsson and D. Parker, J. Chem. Soc., Perkin Trans. 2, 2000, 1819–1831 RSC.
- R. F. H. Viguier and A. N. Hulme, J. Am. Chem. Soc., 2006, 128, 11370–11371 CrossRef CAS.
- A. Thibon and V. C. Pierre, J. Am. Chem. Soc., 2009, 131, 434–435 CrossRef CAS.
- K. Lee, V. Dzubeck, L. Latshaw and J. P. Schneider, J. Am. Chem. Soc., 2004, 126, 13616–13617 CrossRef CAS.
- P. Atkinson, B. S. Murray and D. Parker, Org. Biomol. Chem., 2006, 4, 3166–3171 RSC.
- B. Song, G. Wang, M. Tan and J. Yuan, J. Am. Chem. Soc., 2006, 128, 13442–13450 CrossRef CAS.
- O. V. Vieira, R. J. Botelho and S. Grinstein, Biochem. J., 2002, 366, 689–704 CAS.
- C. P. Montgomery, B. S. Murray, E. J. New, R. Pal and D. Parker, Acc. Chem. Res., 2009, 42, 925–937 CrossRef CAS; E. J. New, D. Parker, D. G. Smith and J. W. Walton, Curr. Opin. Chem. Biol., 2010, 14, 238–246 CrossRef CAS.
- (a) B. S. Murray, E. J. New, R. Pal and D. Parker, Org. Biomol. Chem., 2008, 6, 2085–2094 RSC; (b) C. P. Montgomery, E. J. New, L. O. Palsson, D. Parker, A. S. Batsanov and L. Lamarque, Helv. Chim. Acta, 2009, 92, 2186 CrossRef CAS.
- (a) E. J. New, D. Parker and R. D. Peacock, Dalton Trans., 2009, 672–679 RSC; (b) F. Kielar, G.-L. Law, E. J. New and D. Parker, Org. Biomol. Chem., 2008, 6, 2256–2258 RSC.
- F. Kielar, A. Congreve, G.-L. Law, E. J. New, D. Parker, K.-L. Wong, P. Castreño and J. de Mendoza, Chem. Commun., 2008, 2435–2437 RSC.
- E. J. New and D. Parker, Org. Biomol. Chem., 2009, 7, 851–855 RSC.
- M. O. J. Olson, M. Dundr and A. Szebeni, Trends Cell Biol., 2000, 10, 189–196 CrossRef CAS.
- J. Yu, D. Parker, R. Pal, R. A. Poole and M. J. Cann, J. Am. Chem. Soc., 2006, 128, 2294–2299 CrossRef CAS.
- S. Veldhoen, S. D. Laufer, A. Trampe and T. Restle, Nucleic Acids Res., 2006, 34, 6561–6573 CrossRef CAS.
- L. H. Wang, K. G. Rothberg and R. G. Anderson, J. Cell Biol., 1993, 123, 1107–1117 CrossRef CAS.
- P. A. Orlandi and P. H. Fishman, J. Cell Biol., 1998, 141, 905–915 CrossRef CAS; J. E. Schnitzer, P. Oh, E. Pinney and J. Allard, J. Cell Biol., 1994, 127, 1217–1232 CrossRef CAS.
- A. Arcaro and M. P. Wymann, Biochem. J., 1993, 296, 297–301 CAS.
- P. R. Hoffmann, A. M. deCathelineau, C. A. Ogden, Y. Leverrier, D. L. Bratton, D. L. Daleke, A. J. Ridley, V. A. Fadok and P. M. Henson, J. Cell Biol., 2001, 155, 649–659 CrossRef CAS.
- L. J. Hewlett, A. R. Prescott and C. Watts, J. Cell Biol., 1994, 124, 689–703 CrossRef CAS; H. U. Keller, J. Cell. Physiol., 1990, 145, 465–471 CrossRef CAS.
- H. H. Mollenhauer, D. J. Morré and L. D. Rowe, Biochim. Biophys. Acta, 1990, 1031, 225–246 CAS.
- D. D. Pless and R. B. Wellner, J. Cell. Biochem., 1996, 62, 27–39 CrossRef CAS.
- G. Drin, S. Cottin, E. Blanc and A. R. Rees, J. Biol. Chem., 2003, 278, 31192–31201 CrossRef CAS.
- W. A. Dunn, A. L. Hubbard and N. N. Aronson, Jr., J. Biol. Chem., 1980, 255, 5971–5978 CAS; S. Kessner, A. Krause, U. Rothe and G. Bendas, Biochim. Biophys. Acta, 2001, 1514, 177–190 CAS.
- W. Pendergrass, N. Wolf and M. Poot, Cytometry, Part A, 2004, 61a, 162–169 Search PubMed.
- R. J. Kessler, C. A. Tyson and D. E. Green, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 3141–3145 CrossRef CAS.
- M. G. Ormerod, in Flow Cytometry: A Practical Approach, ed. M. G. Ormerod, Oxford University Press, Oxford, 2000 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section. See DOI: 10.1039/c0sc00105h |
| This journal is © The Royal Society of Chemistry 2010 |