Enlarging the reactive cone of acceptance by exciting the C–H bond in the O(3P) + CHD3 reaction
Fengyan
Wang
and
Kopin
Liu
*
Institute of Atomic and Molecular Sciences (IAMS), Academia Sinica, P.O. Box 23-166, Taipei, Taiwan 10617. E-mail: kliu@po.iams.sinica.edu.tw
First published on 12th May 2010
Abstract
The effects of the CH stretching vibration of methane on the O(3P) + CHD3 reaction are elucidated in a crossed-beam imaging experiment. For the H-atom abstraction channel, the ground state reaction leads mainly to the vibrational ground states of products, OH(v = 0) + CD3(v = 0). When the reactant CHD3 is prepared with one-quantum excitation of CH stretching vibration (v1 = 1), more than 90% of OH coproducts that formed concomitantly with the CD3(v = 0) product are in v = 1. Significant vibrational enhancement in the reaction rate is observed, and the product angular distribution shifts from a backward-dominance in O(3P) + CHD3(v = 0) to a sideways-peaking upon CH stretching excitation. As to the D-atom abstraction channel, the C–H bond excitation, despite being conserved in the CHD2 product, unexpectedly hinders the overall reactivity of the unexcited C–D bond. We interpret the experimental findings as the result of enlarging the reactive cone of acceptance in abstracting the H atom at the expense of the cone for the D-atom transfer channel.
1. Introduction
The reaction of O(3P) + CH4 is a primary step in methane combustion;1 as such, the kinetics of this reaction have been extensively studied experimentally2 and theoretically.3–8 Experimental dynamics studies are, however, sparse. Suzuki and Hirota measured the nascent vibrational distribution in the umbrella mode of CH3,9 and later Sweeney et al. measured the rotational distribution of OH.10 More recently, Minton and co-workers also investigated this reaction, focusing instead on the H + OCH3 product channel at hyperthermal energies.11,12 The excitation function (i.e., the dependence of reaction cross section on collision energy Ec) over the energy range of 1.6–3.2 eV (1 eV = 96.5 kJ mol−1) and the product channel branching (H + OCH3vs. OH + CH3) at Ec = 2.8 eV were reported. All previous experimental studies referred to the reaction with the vibrational ground state of CH4. The only exception is that reported several years ago by this laboratory on the effects of the bending excitations of CD4 and CHD3 on chemical reactivity and other dynamical observables.13 Contrary to the theoretical predictions, little rate-promotion was observed by exciting the bending modes of methane. Moreover, when a bend-excited CD4 or CHD3 does react with O(3P), the initial bending energy, instead of flowing into the umbrella mode of methyl radicals as theories predicted,3,14,15 is preferentially channelled into OD/OH vibrations.The product channel of hydroxyl + methyl radicals is slightly endothermic by 6.3–10.5 kJ mol−1, depending on the isotopic variants of methane. Both thermal kinetics results2 and theoretical calculations4–6 indicated a high barrier, ∼40 kJ mol−1 when the zero point energy is included, to this channel. As the O(3P) atom approaches the CH4 molecule in C3v symmetry (3E potential energy surface, PES), a Jahn–Teller conical intersection occurs.4–6 A non-totally symmetric vibrational mode that shifts the O(3P) atom off the C–H axis splits the 3E PES into the 3A′ and 3A′′ PESs. Ab initio calculations showed that this reaction has a nearly collinear O–H–CH3 transition state due to the conical intersection of the two lowest electronic states. The saddle-point geometry is, however, somewhat uncertain.4–6 The predictions of the relative order of the lengths of the breaking C–H bond and forming O–H bond obtained at various ab initio levels are not consistent. Nonetheless, on the basis of that both the breaking and forming bonds are elongated (roughly 15% to 25% longer than the corresponding equilibrium distances in the OH and CH4 molecules) and the predicted HCH angle is 104° (i.e., lying between the two limiting angles, 109.5° and 90°), the transition state is perhaps best viewed as neither an early (reactant-like structure) nor a late (product-like) barrier, but rather a more or less central barrier.
A number of theoretical dynamics studies using quasiclassical trajectory calculations6,8,16 or reduced-dimensional quantum dynamics,14,15,17–20 mostly on the PES developed by Espinosa-Garcia and Garcia-Bernaldez,5 have been reported. Particularly relevant to the present experiment are the reports focused primarily on the effects of stretching vibration of methane on reactivity.15,17–19 In a pioneering study, Palma and Clary examined the effect of symmetric and antisymmetric stretching excitation of CH4 on O(3P) + CH4.17 Using a four-dimensional quantum scattering model (the system has twelve dimensions), which explicitly accounted for the umbrella, symmetric stretching (s.s.) and antisymmetric stretching (a.s.) vibration of CH4, they found that s.s. vibration is more efficient in promoting reaction than the a.s. mode. Compared to the ground state reaction with an equivalent amount of energy in translation, both stretching vibrations produce a significant enhancement in reactivity. They also found that exciting either stretching mode yields an inverted population in the umbrella vibration of the CH3 product, in contrast to the ground state reaction, for which the population is non-inverted. Similar investigation was reported on the O(3P) + CH3D → OD + CH3 reaction.18 Again, a preferential promotion in reaction rate by s.s. excitation over the translation energy was found, but exciting the a.s. vibration led to a deceleration of the reaction rate. Yu and Nyman15 also studied the quantum dynamics of the O(3P) + CH4 reaction based on a rotating bond umbrella model and reached the same qualitative conclusion on the vibrational enhancement in reactivity as that by Clary. In addition, their results showed that exciting the CH4 stretching mode yields predominantly vibrationally unexcited OH products, though the population in OH(v = 1) is enhanced. More recently, Yang et al. reported a seven-dimensional quantum dynamics study of the O(3P) + CH4 reaction.19 By restricting the non-reacting CH3 group under C3v symmetry and fixing the CH bond length in the CH3 group, the remaining seven degrees of freedom were treated exactly in this model. Although the quantitative results differ from the previous theoretical predictions, the same qualitative conclusions were drawn: The initial CH stretching excitation enhances the reactivity, but only part of vibrational energy can be used to reduce the reaction threshold. Unfortunately, due to the restriction to C3v symmetry, this model cannot compare the mode-specific reactivities between the s.s. and a.s. stretching excitations.
Reported here is the first experimental investigation into the effects of the CH stretching excitation (v1 = 1) on the reactivity of O(3P) + CHD3. The choice of this particular isotopic methane seems intuitively appealing in that one-quantum excitation of the v1 mode in CHD3 is essentially localized in the C–H bond without much mixing from the other modes.21,22 Here, the focus is on the major vibration states of the two isotopic product channels: CD3(v = 0) + OH(v) and CHD2(v1 = 1) + OD(v). In addition to the vibrational enhancement factor, the correlated OH/OD vibrational branching ratios and angular distributions were measured and compared to those in the ground state reactions.
In a broader perspective, the present study is intimately related to our recent reports on the CHD3(v1 = 1) reactions with the F and Cl atoms.23–27 All three reactions proceed via a direct H- or D-atom abstraction pathway. Yet, as depicted in Fig. 1, they are characterized by vastly different energetics and barrier properties. The F-atom reaction is highly exothermic and has a small barrier located in the early part of the entrance valley.28,29 Both Cl- and O(3P)-atom reactions are slightly endothermic with substantially higher barriers; thus, by the Hammond postulate,30 the barriers to reactions are not expected to be late. Ab initio calculations predicted a more product-like transition state structure in the Cl-atom reaction31,32 than the O(3P) case, albeit given the aforementioned uncertainty for the latter reaction.4–6 Hence, a close comparison of the behaviours of three reactions upon CH stretching excitation could shed some light on broad questions: what are the deciding features, in the topography of the multidimensional PES, that govern the observed changes in reactivity, and how?
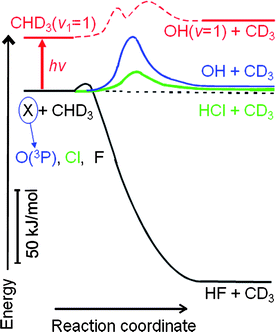 | ||
| Fig. 1 Schematic representation of the potential energy profiles of three direct H-atom abstraction reactions along the reaction coordinate. All energetics are roughly scaled according to the available experimental results, and the locations of the barriers are in keeping with the ab initio calculated saddle point geometries. Note the vastly different heats of reaction, the barrier heights and locations along the reaction coordinate. Also illustrated as the (roughly sketched) dashed red line is the vibrationally excited curve that adiabatically correlates the O(3P) + CHD3(v1 = 1) reactant pair to the OH(v = 1) + CD3(v = 0) product pair. The appearance of a shallow dynamical well in the vicinity of the transition state region arises from the dramatic decrease of the CH stretching frequencies, according to the theoretical prediction (ref. 5). The reduction of the symmetric stretching frequencies along the reaction coordinate also lowers and shifts the excited adiabatic barrier height, which could lead to significant impacts to reaction dynamics (see text). | ||
2. Experimental methods
The experiments were conducted in a crossed molecular beam apparatus equipped with a time-sliced, velocity-mapped ion imaging detector. The detailed setup33 and experimental procedures23–27 have been described previously; only the pertinent features will be mentioned here. Two doubly-skimmed molecular beams, a discharge-generated O-atom beam34 (a mixture of 5% O2 and He at 6 atm; a trace amount of H2 was added to remove the residual F-atom in the beam35) and a seeded CHD3 beam (∼20% in H2 at 5 atm for acceleration), were crossed in a differentially pumped chamber. A tunable infrared (IR) OPO/OPA (optical parametric oscillator/amplifier) excited a fraction of the CHD3 molecules to a single rovibrational state (v1 = 1, j = 2) through a multipass ring reflector36 that was located directly in front of the first skimmer. A home-made photo-acoustic spectrometer aided in tuning the OPO frequency to the desired 110R(1) transition at 3005.57 cm−1.37 After the collisions, the methyl radical products were detected by using a (2 + 1) resonance-enhanced multiphoton ionization (REMPI) scheme near 333 nm via the two-photon excited intermediate 3p-2A′′ Rydberg state.38,39 The state-tagged product velocity distribution was measured by a time-sliced, velocity-imaging technique of the REMPI ions.33 The Q branch of the REMPI band was used to detect the ground vibrational state of CD3(v = 0)38 or CHD2(v1 = 0, 1).39,40 To interrogate the effects of CH stretching excitation, at each Ec the product images were acquired with IR-on and IR-off alternately to minimize the long-term drift and other possible systematic errors. The image accumulation time was typically 1–4 h depending on the individual signal strength.3. Pair-correlated product images
Fig. 2 exemplifies a few raw images of the CD3(v = 0) products at Ec = 31.8 and 47.7 kJ mol−1, respectively. The IR-off image pictures the dynamics of the ground state reaction (excluding the backgrounds), whereas the IR-on image comprises the features from both the stretch-excited reaction and the residual IR-off image due to the reaction with unexcited CHD3 molecules.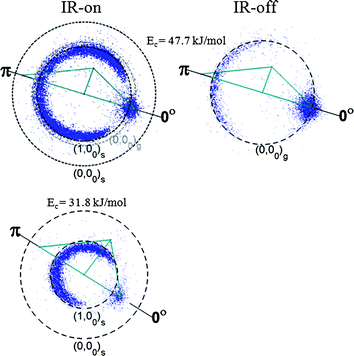 | ||
| Fig. 2 Time-sliced raw images of the probed CD3(v = 0) products from the O(3P) + CHD3 reaction at Ec = 31.8 kJ mol−1 and 47.7 kJ mol−1 for IR-on (left) and IR-off (right). Superimposed on the image is the Newton (reactant velocity vectors) diagram. The forward direction, 00, is defined as the initial CHD3-beam direction in the centre-of-mass frame. The round-shaped feature along 00 is the beam-generated background noise. The ring-like features of the product pairs are assigned as labelled, and the dashed circle indicates the maximal recoil speed of the given product pair. | ||
The IR-on image at Ec = 31.8 kJ mol−1 displays a clear backward/sideways ring that disappeared when the IR laser was blocked. Hence, the ring-like structure arises from the O(3P) + CHD3(v1 = 1) reaction, and the threshold to the ground state reaction is apparently higher than the collisional energy of 31.8 kJ mol−1. At Ec = 47.7 kJ mol−1, the ground state reaction (the IR-off image) is dominated by a backward-peaking ring—a characteristic feature for reactions proceeding via a direct rebound mechanism.30 On energetic grounds (the heat of endothermicity is 8.7 kJ mol−1), this ring can be identified as the formation of the product pair, OH(v = 0) + CD3(v = 0), or for brevity the (0, 00)g pair with the subscript “g” denoting the ground state reaction. The narrowness of the ring structure indicates low rotational excitation of the OH(v = 0) coproducts, in agreement with previous findings.10 For the IR-on image at the same Ec, again a single ring dominates, showing a sideways peaking angular distribution. The vibrational frequencies of the v1-mode of CHD3 and the OH stretch are not very different; thus, the added reactant vibration energy (36.1 kJ mol−1) could largely be balanced by one-quantum excitation of the OH product (42.6 kJ mol−1), yielding a slightly slower recoil velocity than the IR-off image. Hence, the observed single ring on the IR-on image is ascribed to the overlapped contributions from both the stretch-excited and the ground state reactions, labelled as (1, 00)s and (0, 00)g, respectively. [The definition of (1, 00)s is the same as (0, 00)g, except the subscript “s” denotes a stretch-excited reaction.]
To disentangle the overlapped ring features, a precise knowledge about the fraction of excited CHD3 being pumped in the beam and contributing to the observed signals, n≠/n0, is required.24 To this end, we exploited the depletion method developed previously in the F + CHD3(v1 = 1) reaction23 and found typically n≠/n0 ≈ 30% in this study. To analyze the image, the backgrounds were first subtracted to obtain the true IR-on and IR-off images. We then performed the density-to-flux transformation to account for the non-uniform detection sensitivity over the product laboratory velocity.33 By scaling down the IR-off angular distribution to (1 − n≠/n0) and subtracting it from the IR-on distribution, the genuine distribution of the (1, 00)s pair was recovered.24
The resultant pair-correlated angular distributions for (1, 00)s, along with that deduced directly from the IR-off image for (0, 00)g, at the two Ecs are depicted in Fig. 3. The dramatic change in angular distributions upon CH stretching excitation is vividly displayed. Also shown on the left are the CD3(00) product speed distributions. The energetic difference (6.5 kJ mol−1) in producing the (1, 00)s and (0, 00)g pairs from the two respective reactions at Ec = 47.7 kJ mol−1 is clearly manifested as a noticeable peak shift of the two speed distributions. A closer inspection also reveals a small tail at higher speeds, which energetically corresponds to the formation of a (0, 00)s pair in the stretch-excited reaction.
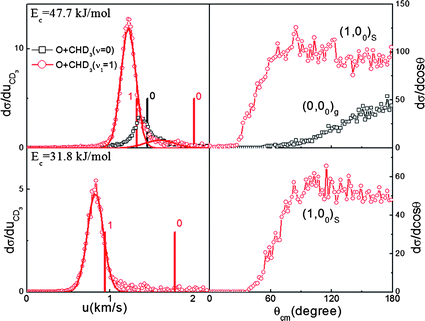 | ||
| Fig. 3 The CD3 product speed distributions (left) and the pair-correlated angular distributions (right) in the ground (in black) and excited states (in red) reactions. The vertical lines mark the CD3 product speed limits for the concomitantly formed OH(v = 0 or 1) coproducts. The solid curves are the simulated speed distributions. | ||
At a fixed Ec the IR-on and IR-off images were experimentally normalized to each other, so that the resultant product speed and angular distributions for the stretch-excited and ground-state reactions are also normalized. It is then straightforward to derive the vibrational enhancement factor σs/σg and the vibrational branching ratio. Performing the same imaging measurements at other Ecs and normalizing them according to the procedure detailed previously,23,26,41 the reactive excitation function can then be determined.
4. Vibrationally hot OH from CH stretching excitation
Fig. 4 summarizes the results. We will first examine the energy disposal and the correlated vibrational branching ratio, OH(v = 1)/OH(v = 0). In the ground state reaction, only OH(v = 0) was observed, in accord with energetic expectation that the formation of the (1, 00)g product pair requires at least 51.3 kJ mol−1 of energy. Upon CH stretching excitation, the additional vibrational energy (36.1 kJ mol−1) lowers the energetic threshold for (1, 00)s down to 15.2 kJ mol−1. Experimentally, the observed OH vibrational distribution (Fig. 4(b)) is highly inverted, in sharp contrast to the theoretical prediction.15 In addition, such an inverted distribution shows little dependence on collisional energies. The observation of a highly inverted OH vibration distribution differs vastly from a simpler benchmark reaction of O(3P) + H2(v = 1), for which two bulb experiments at 300 K using different ways of generating O(3P) atoms have been reported.42,43 At a mean collision energy of 4 kJ mol−1, the ratio of OH(v = 1)/OH(v = 0) was estimated to be <0.04.42 For a translationally hot O(3P) atom at higher average Ec ∼ 20 kJ mol−1, the ratio increased to about 2.1.43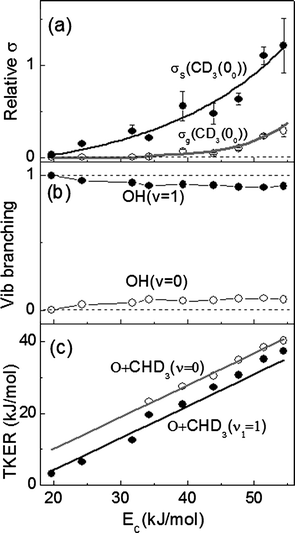 | ||
| Fig. 4 (a) The normalized excitation functions for the ground state (σg) and the stretch-excited (σs) reactions as the major methyl product CD3(v = 0) was probed. The error denotes one standard deviation. The lines are visual guides. (b) The OH vibrational branching fraction, typical error of ±3%, in the O(3P) + CHD3(v1 = 1) → OH(v) + CD3(00) reaction. (c) Dependence of total kinetic energy release (TKER) on Ec for the ground state (○) and stretch-excited (●) reactions. Two straight lines are the respective predictions based on a kinematic model (see text). | ||
Intuitively, the observation of vibrationally hot OH appears in line with the vibrationally adiabatic picture.26,27,30 As illustrated in Fig. 1, in the vibrationally adiabatic framework, the initial CH-stretching mode in the O(3P) + CHD3(v1 = 1) reaction is adiabatically correlated to the OH(v = 1) + CD3(00) product pair (or alternatively to the OD(v = 0) + CHD2(v1 = 1) pair in the D-atom transfer channel). If the vibrationally nonadiabatic couplings of the reactant v1-mode to the reaction path is small, the adiabatic picture would lead to an inverted OH vibrational distribution. However, ab initio calculations on the O(3P) + CH4 reaction4–6 showed a significant reduction of the s.s. vibrational frequency (or the CH stretching mode in the case of CHD3) in the transition state region, indicative of a non-negligible curvature coupling5 to the motion along the reaction path. In addition to this nonadiabatic coupling en route to the barrier, ab initio calculation further predicted a second nonadiabatic coupling on the product side of the saddle point, which corresponds primarily to the curvature coupling between the reaction path and the OH stretching motion.5
Taking the ab initio results into consideration, a simple and intuitive picture emerges. As the O(3P) + CHD3(v1 = 1) colliding pair approaches the first curvature coupling region, nonadiabatic transitions take place by funnelling the reactant CH vibrational energy into the reaction coordinate and the reactive trajectories bifurcate.26,27 Those with the initial vibrational excitation retained in the O–H–C moiety will stay adiabatically and eventually lead to the OH(v = 1) + CD3(00) product pair. The other pathway mediated via the nonadiabatic transition will proceed over the ground vibrational surface and encounter the second curvature coupling that allows the energy in the reaction coordinate to flow back to the orthogonal vibration modes, now the stretch of the OH-moiety. The fact that more than 90% of the OH products are born in v = 1 must then imply either a rather small nonadiabatic transition probability by the first curvature coupling or an extremely efficient second nonadiabatic transition. Further theoretical investigations will be needed in this regard for deeper insights.
Alternatively, both the low OH rotational excitation (inferred from the narrowness of product speed distributions, Fig. 3) and the inverted OH vibrational population suggest a kinematic origin.30 This is a light-atom transfer reaction; thus, treating the CD3-moiety as a pseudo-atom, the small skew angle (19.2°) ensures that the reactant vibrational coordinate is nearly parallel with that of the product in the mass-scaled coordinate system, rendering the efficient transfer of reactant into product vibrational excitation. This simple kinematic consideration predicts that the average product kinetic energy release <ET> in the ground state reaction should scale as 〈ET〉 = (Ec − ΔHrx)cos2β, where ΔHrx is the endothermicity, and the skew angle is given by β = cos−1[mAmC/(mA+mB)(mB + mC)]1/2 for the A + BC → AB + C reaction.30 As shown in Fig. 4(c), the excellent agreement between the experimental data for O(3P) + CHD3(v = 0) and the model prediction (the grey line), in which no adjustable parameter is invoked, strongly supports the role of kinematics in governing this dynamical attribute.
Application of this simple kinematic model to the stretch-excited reaction is not without ambiguity due to the uncertainty in quantifying the disposal of the reactant vibration energy. In keeping with the aforementioned reasoning on a facile vibrational energy transfer between reactant and product, we treated Ec in the above equation as the reactant total energy and likewise, ΔHrx as the energetics for forming the OH(v = 1) + CD3(00) products. The predicted kinetic energy release in the stretch-excited reaction is then depicted as the black line in Fig. 4(c). As is seen, the Ec-dependence of the experimental data is reasonably (or fortuitously) reproduced. In that regard, we noted that a previous reduced dimensionality quantum dynamics study did not yield highly inverted OH vibrational distributions in O(3P) + stretch-excited CH4,15 implying that, besides the kinematic constraint, other dynamics factors must be at work. Moreover, for a kinematically similar reaction Cl + CHD3, the recent studies showed that although the vibrational branching fraction, HCl(v = 1)/HCl(v = 0, 1), does increase from 0.02 in the ground state reaction to 0.44 in Cl + CHD3(v1 = 1),26,27 the quantitative results are not fully compatible with the above simple kinematic arguments. In any event, two complementary viewpoints are presented here. While both may be important ingredients, they do not seem to provide a full picture of the underlying mechanism for such a highly inverted OH vibration distribution.
5. Mechanistic origin of the vibrational effect on reaction rate
5.1 The OH + CD3(v = 0) channel
Turning to the reactive excitation function shown in Fig. 4(a), we first note that the ground state reaction threshold occurs around 33.5 kJ mol−1, which is lower than the ab initio predicted barrier height of 40 kJ mol−1,4–6 indicative of significant tunneling effects. Exciting the C–H bond in CHD3 lowers the reaction threshold to about 19 kJ mol−1, so only about 40% of the vibrational energy deposited can be used to reduce the reaction threshold. In the vibrationally adiabatic framework, the reduction of the reaction threshold could originate from the abrupt decrease of the CH stretching frequency en route to the barrier,4–6 yielding a lower barrier to reaction on the vibrationally adiabatic excited surface, as illustrated in Fig. 1. Also notable is the marked increase in reaction cross section with the CH stretch-excited reactant at a fixed Ec. The enormous enhancement at lower Ec is simply due to the different thresholds of the two reactions; at higher Ec > 45 kJ mol−1, the ratios of σs/σg level off, approaching a factor of about five.It is instructive to contrast these results with the analogous reactions of F and Cl atoms with CHD3(v1 = 1).23–27 In the case of the F-atom reaction,23 exciting the CH stretch in CHD3 inhibits the C–H bond cleavage, resulting in a deceleration of overall reaction rate. The reaction threshold for Cl + CHD3(v = 0) → HCl + CD3(00) was previously measured to be about 10 kJ mol−1.44 Exciting the CH stretching mode reduces the threshold to Ec < 2 kJ mol−1 and the vibrational enhancement factor at a fixed Ec approaches a value of two at higher Ecs.26 Hence, a comparably higher rate-promotion factor is observed here for the O(3P) reaction than the Cl, which is counterintuitive. Referring to Fig. 1 and as alluded to earlier, the transition-state structure in Cl + CHD3 is closer to being product-like than that in the O(3P) reaction. One cannot therefore interpret, by Polanyi's rules,30,45 the larger vibrational enhancement for O(3P) as due to a barrier to reaction located closer to the exit valley. What could then be the origin?
One way to think of the rate promotion is in terms of the opacity function P(b),30 which is the reaction probability P as a function of the impact parameter b. There are in general two effects to account for rate promotion of a reaction. The first is the increase of the probability over a limited b-range and the other is the extension of the b-range for reaction. The impact parameters in a direct reaction are closely correlated—at least classically—to the product scattered angles: the smaller b leads to the larger scattering angle and vice versa.30 Therefore, the product angular distribution dσ/d(cosθ) could provide a clue to differentiate the two effects. As shown in Fig. 3, the angular distribution of the (0, 00)g product pair at Ec = 47.7 kJ mol−1 is entirely confined within the backward hemisphere and peaks at 180°, indicating the dominance of small impact parameter collisions for the ground state reaction. For the stretch-excited reaction of O(3P) + CHD3(v = 1), the angular distribution of the major product pair (1, 00)s protrudes significantly into the sideways and forward hemisphere, demonstrating a substantial extension of the range of reactive impact parameters, thus, the change of the opacity function.
Previously, we illustrated in several reaction systems46–49 that an examination of the evolution of pair-correlated angular distributions dσ/d(cosθ) against the collision energy Ec and the scattering angle θ could be particularly illuminating to provide a global view of the underlying reaction mechanism. Fig. 5 summarizes such results for O(3P) + CHD3(v = 0) → OH(v = 0) + CD3(00) and O(3P) + CHD3(v1 = 1) → OH(v = 1) + CD3(00). The pattern seen in the ground state reaction—angular distributions peaking at 180° and confined within the backward hemisphere—features the typical characteristics of a direct rebound mechanism governed by small impact-parameter collisions. The evolution of the angular distribution with Ec further suggests that the increase of the ground state reactivity with Ec over the energy range of this study, Fig. 4(a), is due primarily to the increase of the reaction probability over a limited b-range. For the stretch-excited reaction the angular distributions remain backward at lower Ec, but with the increase in Ec an additional feature partakes, which gradually protrudes into the forward hemisphere yet with a clear cut-off against the small angles. The behaviour of this additional feature is reminiscent of a peripheral reaction mechanism demonstrated previously in Cl + methane,44,50,51 for which the reaction favors large impact-parameter collisions. The fact that as the collision energy increases, the product angular distribution of the stretch-excited reaction not only continuously extends its angular range (i.e., extending the b-range) but also displays a clear preference towards the sideways/forward direction appears in line with the notion of a wider range of the orientation of the attacked C–H bond for collisions at a fixed (large) impact parameter.52
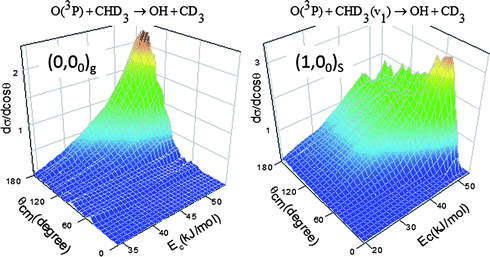 | ||
| Fig. 5 Three-dimensional plot of dσ/d(cosθ) against θ and Ec, which summarizes the evolution of the product angular distributions with the increase in collision energy, thus the global reaction dynamics. Note the distinct patterns of the two reactions. | ||
Two decades ago, Levine et al. proposed a mechanism for vibrationally enhanced reactivity in several atom + diatom reactions.53,54 The rate promotion is attributed to the opening up of the cone of acceptance upon reactant vibrational excitation, thereby, extending the range of reactive impact parameters. They further pointed out that such a steric effect could be particularly prominent for nearly thermoneutral reactions.53 As just elucidated, the global features of two distinctive patterns displayed in Fig. 5—changing from a backward-peaking pattern in the ground state reaction to a sideways-dominant one upon CH stretching excitation—are therefore entirely conformable with Levine's mechanism of enlarging the reactive cone of acceptance. We surmise that the driving force behind this vibrationally-induced steric effect stems from the topography of long-range anisotropic interactions in the entrance valley, which must change in such a way as to orient the reactants and pull the trajectories towards the transition state. Remarkably, this conjectured focusing-effect appears precisely opposite to the steric effect observed recently in F + CHD3(v1 = 1),23 for which the prohibition of C–H bond rupture (to form HF) upon CH stretching excitation was ascribed to steering or deflecting the approaching F atom away from the attacked H atom.23,55
5.2 The OD + CHD2(v1 = 0, 1) channels
One may ask: how does the enlarged reactive cone of acceptance toward the stretch-excited C–H bond influence the other isotopic channel, OD + CHD2? In the previous study of F + CHD3(v1 = 1) an enhanced production of CHD2(v1 = 1) was found,23 which is anticipated by the spectator-bond model. Unexpectedly, the initial CH stretching excitation of CHD3 exerts negative impacts on the D-atom transfer channel by slowing down the total formation rate of DF + CHD2. As for the Cl + CHD3(v1 = 1) case, the initial C–H bond excitation does not affect the overall rate of formation of DCl, but rather yields a vastly different CHD2 state distribution.27Fig. 6 presents two images of the present reaction when the product states CHD2(v = 0) and CHD2(v1 = 1) were probed at Ec = 51.5 kJ mol−1.56 The ground state CHD2 image was acquired without IR irradiation because an identical IR-on image was obtained, except for the intensity being attenuated. Evidently, the yield of the CHD2(v = 0) product in the O(3P) + CHD3(v1 = 1) reaction is at most a small fraction of that in the ground state reaction, similar to the previous finding in F + CHD3(v1 = 1).23 On energetic grounds (the endothermicity of this isotope channel is 9.4 kJ mol−1), the two ring-like features can be identified as the correlated OD(v = 0) and OD(v = 1) states for the outer (the faster speed) and the inner features, respectively.
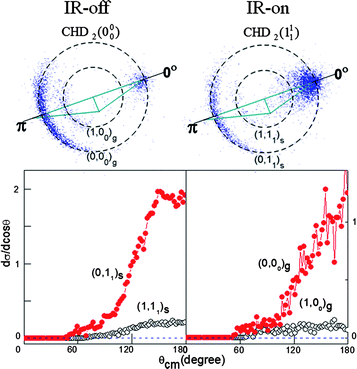 | ||
| Fig. 6 Two raw images of the probed CHD2(v1 = 0, 1) products. The REMPI bands used are indicated. Also shown are the product pairs assigned to the ring features. The lower panel presents the corresponding angular distributions. | ||
Shown on the right is the IR-on image for CHD2(v1 = 1); no CHD2(v1 = 1) products can be detected in the ground state reaction, even though it is energetically feasible. The appearances, in all aspects, of the two images (Fig. 6) are nearly indistinguishable. Since the vibrational energy (37.2 kJ mol−1) of the probed state CHD2(v1 = 1) contains almost the same energy as that (36.1 kJ mol−1) deposited into the reactant CHD3(v1 = 1), the observed features are assigned to the (v = 0) and (v = 1) states of the OD coproducts in both reactions. Quantitative analysis revealed that the vibrational branching ratios OD(v = 1)/OD(v = 0) are indeed nearly the same, 0.21 and 0.19 in the two respective reactions of O(3P) + CHD3(v = 0) → OD(v) + CHD2(00) and O(3P) + CHD3(v1 = 1) → OD(v) + CHD2(11).57 Also shown in Fig. 6 are the corresponding angular distributions of the product pairs. Again, the distributions are alike—though the dominant (0, 11)s pair seems a bit more sharply peaking at 180° than (0, 00)g. All these results then imply similar pathways of the two reactions, in accord with the intuitive spectator-bond picture.
However, the signal strength for probing the CHD2(11) product in the reaction with CHD3(v1 = 1) was significantly weaker than that for CHD2(00) from O(3P) + CHD3(v = 0). After taking into account the n≠/n0 (∼ 30%) factor and the relative detection sensitivity of the REMPI bands (∼1.9 in favor of CHD2(111)),58 we found that σs(11)/σg(00) is merely 0.2. Since CHD2(11) turns out to be the only state with detectable enhancement in this isotopic product channel, we therefore concluded that the D-atom abstraction reactivity is substantially suppressed upon CH stretching excitation of CHD3. This conclusion goes against the spectator paradigm, despite the fact that the initially deposited energy remains intact in the product C–H bond and that all observed dynamical attributes of the CHD2(11) product in the stretch-excited reaction broadly resemble those of CHD2(00) in the ground state reaction.59 To reconcile these seemly conflicting observations, we conjecture that the opening up of the cone of acceptance toward the stretching C–H bond is at the expense of those trajectories that otherwise will abstract the D atoms, i.e., by narrowing the cone of reaction of the D-atoms.
6. Conclusions
Both the integral and differential cross sections in reactions of O(3P) with the ground state and CH stretch-excited CHD3 reactants are reported here over the energy range of chemical interest. The ground state reaction producing OH + CD3 has a threshold at ∼33.5 kJ mol−1, and the dynamics are characterized by the rebound mechanism. Exciting the C–H bond of the CHD3 reactant lowers the threshold to about 19 kJ mol−1 and promotes the reaction rate. The predominant OH product is vibrationally excited v = 1 and the angular distribution shifts into the forward hemisphere. We interpret these results as an enlarged reactive cone of acceptance upon C–H bond excitation—a vibrationally-induced steric mechanism originally proposed by Levine in atom + diatom reactions. Examining the other isotope channel, OD + CHD2, reveals that the production of CHD2(v = 0) becomes depleted by reactant stretching excitation, whereas the CHD2(v1 = 1) yield is enhanced with its dynamical attributes being similar to that of CHD2(v = 0) in the ground state reaction; though some subtle differences were noted.59 Nevertheless, a negative impact on the D-atom transfer channel, namely slowing down its total reaction rate was observed by CH stretching excitation of CHD3. To comprehend the observations for both isotopic channels, we propose that the enlarged reactive cone of acceptance towards the stretching C–H bond is likely accompanied by narrowing of the cone of acceptance towards the unexcited C–D bonds. Unfortunately, the relative REMPI detection sensitivities of probing the CD3(v = 0) and CHD2(v1 = 0, 1) radical products are yet to be determined (an experimentally challenging task), it remains to be seen if the enlarged cone of acceptance toward the stretching C–H bond could be evenly balanced by the steric hindrance of abstracting the unexcited D-atoms. In other words, will the total reactivity, when considering both isotopic channels, change upon CH stretching excitation? Works to address this issue are under way.Acknowledgements
The authors are indebted to Jui-San Lin and Huilin Pan for their assistance in experiments. Financial support from the National Science Council of Taiwan, Academia Sinica, and the Air Force Office of Scientific Research (grant No. AOARD-09-4030) is gratefully acknowledged.Notes and references
- I. M. Campbell, Energy and the Atmosphere, John Wiley & Sons Ltd., London, 1977 Search PubMed.
- D. L. Baulch, C. J. Cobos, R. A. Cox, C. Esser, P. Frank, Th. Just, J. A. Kerr, M. J. Pilling, J. Troe, R. W. Walker and J. Warnatz, J. Phys. Chem. Ref. Data, 1992, 21, 411 CAS ; and references therein.
- D. C. Clary, Phys. Chem. Chem. Phys., 1999, 1, 1173 RSC.
- J. C. Corchado, J. Espinosa-Garcia, O. Roberto-Neto, Y.-Y. Chuang and D. G. Truhlar, J. Phys. Chem. A, 1998, 102, 4899 CrossRef CAS.
- J. Espinosa-Garcia and J. C. Garcia-Bernaldez, Phys. Chem. Chem. Phys., 2000, 2, 2345 RSC.
- M. Gonzalez, J. Hernando, J. Millan and R. Sayos, J. Chem. Phys., 1999, 110, 7326 CrossRef CAS.
- F. Huarte-Larranaga and U. Manthe, J. Chem. Phys., 2002, 117, 4635 CrossRef CAS.
- D. Troya and E. Garcia-Molina, J. Phys. Chem. A, 2005, 109, 3015 CrossRef CAS.
- T. Suzuki and E. Hirota, J. Chem. Phys., 1993, 98, 2387 CrossRef CAS.
- G. M. Sweeney, A. Watson and K. G. McKendrick, J. Chem. Phys., 1997, 106, 9172 CrossRef CAS.
- D. J. Garton, T. K. Minton, D. Troya, R. Pascual and G. C. Schatz, J. Phys. Chem. A, 2003, 107, 4583 CrossRef CAS.
- D. Troya, G. C. Schatz, D. J. Garton, A. L. Brunsvold and T. K. Minton, J. Chem. Phys., 2004, 120, 731 CrossRef CAS.
- B. Zhang and K. Liu, J. Phys. Chem. A, 2005, 109, 6791 CrossRef CAS.
- J. Palma and D. C. Clary, J. Chem. Phys., 2000, 112, 1859 CrossRef CAS.
- H.-G. Yu and G. Nyman, J. Chem. Phys., 2000, 112, 238 CrossRef CAS.
- R. Sayos, J. Hernando, M. P. Puyudo, P. A. Enriquez and M. Gonzalez, Chem. Phys. Lett., 2001, 341, 608 CrossRef CAS.
- J. Palma and D. C. Clary, Phys. Chem. Chem. Phys., 2000, 2, 4105 RSC.
- J. Palma, J. Echave and D. C. Clary, Chem. Phys. Lett., 2002, 363, 529 CrossRef CAS.
- M. Yang, S.-Y. Lee and D. H. Zhang, J. Chem. Phys., 2007, 126, 064303 CrossRef.
- Q. Cui, M.-L. Wang and J. Z. H. Zhang, Chem. Phys. Lett., 2005, 410, 115 CrossRef CAS.
- X.-G. Wang and E. L. Sibert III, J. Chem. Phys., 1999, 111, 4510 CrossRef CAS.
- S. Carter, H. M. Shnider and J. M. Bowman, J. Chem. Phys., 1999, 110, 8417 CrossRef CAS.
- W. Zhang, H. Kawamata and K. Liu, Science, 2009, 325, 303 CrossRef CAS.
- S. Yan, Y.-T. Wu and K. Liu, Phys. Chem. Chem. Phys., 2007, 9, 250 RSC.
- S. Yan and K. Liu, Chin. J. Chem. Phys., 2007, 20, 333 CrossRef CAS.
- S. Yan, Y.-T. Wu, B. Zhang, X.-F. Yue and K. Liu, Science, 2007, 316, 1723 CrossRef CAS.
- S. Yan, Y.-T. Wu and K. Liu, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12667 CrossRef CAS.
- G. Czako, B. C. Shepler, B. J. Braams and J. M. Bowman, J. Chem. Phys., 2009, 130, 084301 CrossRef.
- J. Zhou, J. J. Lin and K. Liu, J. Chem. Phys., 2004, 121, 813 CrossRef CAS.
- R. D. Levine and R. B. Bernstein, Molecular Reaction Dynamics and Chemical Reactivity, Oxford University Press, New York, 1987 Search PubMed.
- W. T. Duncan and T. N. Truong, J. Chem. Phys., 1995, 103, 9642 CrossRef CAS.
- J. C. Corchado, D. G. Truhlar and J. Espinosa-Garcia, J. Chem. Phys., 2000, 112, 9375 CrossRef CAS.
- J. J. Lin, J. Zhou, W. Shiu and K. Liu, Rev. Sci. Instrum., 2003, 74, 2495 CrossRef CAS.
- I. C. Lu, W.-J. Huang, C. Chaudhuri, W.-K. Chen and S.-H. Lee, Rev. Sci. Instrum., 2007, 78, 083103 CrossRef.
- To ensure a known value of n≠/n0 and to optimize the experimental conditions, the depletion measurement that employed the F + CHD3 reaction (ref. 23) was performed in each day’s experiments. The image acquired with only O2 mixed in He for generating the O-atom beam revealed some faint ring features mostly in the forward direction. The forward and backward rings are, however, not concentric, indicating that they are from two different reactions. As it turned out, the forward feature is the result of the reaction of CHD3 with the residual F-atoms in the beam, whose reaction rate with CHD3 is almost 1000-fold faster than O(3P). The addition of H2 molecules was an effective way to react, thus to consume the contaminated F-atoms through collisions in the supersonic expansion.
- J. Riedel, S. Yan, H. Kawamata and K. Liu, Rev. Sci. Instrum., 2008, 79, 033105 CrossRef.
- D. G. Rea and H. W. Thompson, Trans. Faraday Soc., 1956, 52, 1304 RSC . The band notation 110 designates the spectroscopic transition involving the v1 vibrational mode with zero quantum excitation in the ground state (the subscript) and one quantum in the excited state (the superscript).
- J. Zhou, J. J. Lin, W. Shiu, S.-C. Pu and K. Liu, J. Chem. Phys., 2003, 119, 2538 CrossRef CAS.
- J. Zhou, J. J. Lin and K. Liu, J. Chem. Phys., 2003, 119, 8289 CrossRef CAS.
- B. Zhang, S. Yan and K. Liu, J. Phys. Chem. A, 2007, 111, 9263 CrossRef CAS.
- W. Shiu, J. J. Lin, K. Liu, M. Wu and D. H. Parker, J. Chem. Phys., 2004, 120, 117 CrossRef CAS.
- J. Han, X. Chen and B. R. Weiner, Chem. Phys. Lett., 2000, 332, 243 CrossRef CAS.
- G. C. Light, J. Chem. Phys., 1978, 68, 2831 CrossRef CAS.
- G. Nyman, J. Zhou, B. Zhang and K. Liu, Israel J. Chem., 2007, 47, 1 CrossRef CAS.
- J. C. Polanyi, Science, 1987, 236, 680 CrossRef CAS.
- R. T. Skodje, D. Skouteris, D. E. Manolopoulos, S.-H. Lee, F. Dong and K. Liu, Phys. Rev. Lett., 2000, 85, 1206 CrossRef CAS.
- K. Liu, Annu. Rev. Phys. Chem., 2001, 52, 139 CrossRef CAS.
- K. Liu, J. Chem. Phys., 2006, 125, 132307 CrossRef.
- K. Liu, Phys. Chem. Chem. Phys., 2007, 9, 17 RSC.
- J. Zhou, B. Zhang, J. J. Lin and K. Liu, Mol. Phys., 2005, 103, 1757 CAS.
- B. Zhang and K. Liu, J. Chem. Phys., 2005, 122, 101102 CrossRef.
- We are speaking of P(b,γ), with γ being the angle of attack. Interested readers are referred to ref. 30 and 53 for more detailed discussion of its concept.
- R. D. Levine, J. Phys. Chem., 1990, 94, 8872 CrossRef CAS.
- H. Kornweitz, A. Persky and R. D. Levine, J. Phys. Chem., 1991, 95, 1621 CrossRef CAS.
- G. Czako and J. M. Bowman, J. Am. Chem. Soc., 2009, 131, 17534 CrossRef CAS.
- The signal for the CHD2 product channel was much weaker than that for CD3, so only limited experiments were performed.
- We note that for the D-atom abstraction channel, the initial excitation of CHD3(v1 = 1) will be adiabatically correlated to the OD(v = 0) + CHD2(v1 = 1) pair. If the second nonadiabatic coupling mentioned in Section 4 were at work, then conceivably some energy stored in the reaction coordinate could have flowed into the OD moiety, yielding significantly higher OD(v = 1) branching in the stretch-excited reaction than the observed; and one would not have observed virtually identical IR-on and IR-off images when probing CHD2(v = 0).
- W. Zhang, H. Kawamata, A. J. Merer and K. Liu, J. Phys. Chem. A, 2009, 113, 13133 CrossRef CAS.
- At lower collision energy, Ec = 47.7 kJ mol−1, σs(11)/σg(00) drops to 0.09, and the vibrational branching ratios OD(v = 1)/OD(v = 0) also decrease to 0.11 and ∼0 for the ground state and stretch-excited reactions, respectively. All product angular distributions remain backward-dominant, similar to those shown in Fig. 6. Owing to the much weaker signals, these lower energy results are preliminary.
| This journal is © The Royal Society of Chemistry 2010 |