Asymmetric Brønsted acid catalysis in aqueous solution†
Magnus
Rueping
* and
Thomas
Theissmann
Institute of Organic Chemistry, RWTH-Aachen University, 52074, Aachen, Germany. E-mail: magnus.rueping@rwth-aachen.de; Fax: (+49) 241-809-2665; Tel: (+49) 241-809-94686
First published on 23rd July 2010
Abstract
A biologically inspired reaction design leads to the development of the first highly enantioselective Brønsted acid catalysed reaction in aqueous solution.
For a long time the use of water as a reaction medium in asymmetric catalysis has remained in the shadows. Often feared as a contaminant, water-free systems are frequently given priority. In response to Breslow's work, in which the positive effect of water on the reactivity and selectivity in Diels–Alder reactions was proven, the situation has changed significantly.1 Multiple asymmetric metal catalysed reactions conducted in the presence of water have subsequently been developed.2 In addition to the solvent properties of water, significant industrial aspects come to the fore. Water is cheap, non-toxic, neither flammable nor explosive and has a high specific heat capacity which negates overheating in large industrial processes.
In asymmetric organocatalysis water has now achieved the status of being a topic for scientific discussion.3 However, the great potential of water as a reaction medium, as well as the need for optimized processes, remains undisputed. In the field of asymmetric organocatalysis only chiral amines, primarily proline and its derivatives, have excelled as potent Lewis base catalysts.4,5 These activate substrates through covalent bonds by iminium6a or enamine catalysis.6b
Water as a reaction medium is unknown in asymmetric Brønsted acid catalysis. Ding and co-workers developed a Brønsted acid catalyzed Baeyer–Villiger reaction in chlorinated solvent employing aqueous hydrogen peroxide as reactant.7a,b Furthermore, Schneider et al. reported Brønsted acid catalyzed vinylogous Mannich reactions in which addition of one equivalent of water proved to be beneficial.7c–e Additionally, Schreiner and co-workers were recently able to demonstrate for the first time that activation via hydrogen-bond formation in the presence of water is possible despite water itself being an excellent H-bond donor/acceptor.8 This observation can be explained by hydrophobic effects.9 The non-covalent interactions are not based on the direct attractive intermolecular effects of the reactants but rather on the distinctive necessity of the water molecules to interact with one another.
The formation of such hydrogen bonds results in the non-polar components being aligned such that the contact surface between these molecules and water is minimized (hydrophobic hydration).
Complexing of the non-polar substances gives rise to increased structuring of the surrounding water molecules which is why entropic aspects as well as enthalpic aspects are the driving force behind the hydrophobic hydration.10 The result is an increase in reactant concentration which can accelerate the reaction rate compared to organic solvents.11
The principle of hydrophobic interaction plays an important role in many in vivo processes: enzyme–substrate interactions, protein folding and the formation of lipids in biomembranes are all controlled in such a manner.
The example of glutamate dehydrogenase (GDH) clearly illustrates the principle.12 This ubiquitous enzyme catalyses the transformation of 2-ketoglutarate with ammonia in the presence of nicotinamide adenine dinucleotide (NADH) to form L-glutamate. By changing conformation the active site of the enzyme is opened or closed enabling the reductive amination to occur under hydrophobic conditions.13 In the transition state the 2-imino glutarate is protonated by the aspartate 165 and a chiral iminium ion pair is formed. The iminium ion is then selectively hydrogenated under the hydrophobic conditions instead of being hydrolyzed.
The transfer of this enzymatic process to asymmetric Brønsted acid catalysis using chiral phosphoric acid diesters14 and Hantzsch dihydropyridine as a reductive source has been reported.15 However, these biomimetic systems are limited by the solvent choice. In order to “imitate” hydrophobic effects dry, non-polar solvents, chlorinated and aromatic solvents in particular, are required.
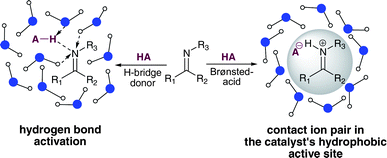
This is due to the competition between water as a strong hydrogen bond donor/acceptor and the chiral catalyst that induces selectivity through non-covalent interactions (Fig. 1, left). In order to resolve this conflict a strongly protected, chiral contact ion pair needs to be formed between the catalyst and the substrate in which the Coulombic interactions can overcome the competitive influence of water (Fig. 1, right).
 | ||
| Fig. 1 | ||
Furthermore, diametrical polarities of the ion pair and the reaction medium are required in order to achieve hydrophobic hydration and to avoid weakening of the contact ion pair. Hence, on the one hand the most non-polar reaction partners and catalysts are required and on the other hand the addition of inorganic salts to the aqueous reaction medium is needed in order to increase the polarity1,16 Therefore, we assumed that under these conditions an enantioselective Brønsted acid catalysed reaction in the presence of water should be possible. Such a reaction would be the first organocatalytic example of a non-covalent asymmetric induction using water as a reaction medium.
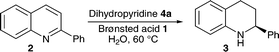
The enantioselective Brønsted acid catalysed transfer hydrogenation of quinolines served as the starting point for our studies. Initial experiments examined the influence of water on the reactivity and enantioselectivity of this reaction as previously only non-polar aprotic, aromatic or chlorinated solvents have been used in Brønsted acid catalysis.
It was assumed that under aqueous reaction conditions the proton transfer from catalyst to the solvent would occur, resulting in non-specific activation of the substrate and leading to greatly reduced enantioselectivity. Therefore, in the first experiment the transfer hydrogenation was conducted in a two phase reaction comprising toluene and water (Table 1, entry 1). From our previous work it was known that the reduction proceeds with an enantiomeric excess of 97% ee in dry toluene.17 Therefore, the 82% ee observed when water was introduced confirmed our assumption that the selectivity would be reduced yet the optical purity of the product was much higher than expected.
|
|
|||
|---|---|---|---|
| Entrya | Solvent | Yield (%)b | ee (%)c |
| a Reaction conditions: quinoline 2, Hantzsch dihydropyridine 4a (2.4 equiv.) and 2 mol% 1e, at 60 °C. b Isolated yields after column chromatography. c Determined by HPLC (Chiralcel OD-H). | |||
| 1 | Toluene–H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
88 | 82 |
| 2 | Dioxane–H2O (1:1) |
65 | 44 |
| 3 | Dioxane | 74 | 97 |
| 4 | H2O, dist. | 81 | 41 |
| 5 | H2O, demin. | 77 | 42 |
| 6 | H2O, NaCl(sat.) (brine) | 78 | 50 |
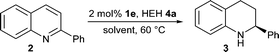
In a comparative study a homogeneous mixture of dioxane and water was applied under identical reaction conditions, as well as a control reaction using dry dioxane (Table 1, entries 2–3). In these one phase reactions the negative effect of water, compared to the aprotic variant, was increased. Therefore, it was particularly gratifying to observe that in pure water reactivity and even moderate enantioselectivity was observed, which confirms the hydrophobic hydration of the chiral ion pair (Table 1, entries 4–6). Discernibly better enantioselectivities were seen when saturated NaCl solution was used as a reaction medium, which is consistent with earlier observations (Table 1, entry 6).
In subsequent experiments we focused on the catalyst structure (Table 2). The highest selectivities were achieved with the sterically demanding catalyst 1c which gave the product in 81% ee (Table 2, entry 3).
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | Yield (%)b | ee (%)c | ||
| a Reaction conditions: quinoline 2, Hantzsch dihydropyridine 4a (2.4 equiv.) and 2 mol% 1, at 60 °C. b Isolated yields after column chromatography. c Determined by HPLC (Chiralcel OD-H). | |||||
|
1 | 1a | [H8]-SiPh3 | 28 | 21 |
| 2 | 1b | 9-Anthracenyl | 81 | 61 | |
| 3 | 1c | 2,4,6-(iPr)3-Ph | 66 | 81 | |
| 4 | 1d | 3,5-(CF3)2-Ph | 79 | 3 | |
| 5 | 1e | 9-Phenanthryl | 74 | 42 | |
In a further step the hydride source was optimized. Various substituted Hantzsch ester derivatives 4 were applied (Table 3) and use of the sterically demanding dihydropyridine 4c provided the best selectivities (Table 3, entry 3).
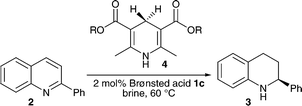
In order to further optimize the reaction parameters the catalyst loading, temperature and concentration were examined. In the latter the concentration of NaCl solution was varied in the range of 0.05–0.5 molar and it was noted that with decreasing concentration higher yields could be isolated. However, a notable influence on the enantioselectivity was not observed. A similar observation was made with the temperature: over a range from 40 to 70 °C almost constant optical purity of the isolated tetrahydroquinolines (89–92% ee) was achieved.
With these results in hand the substrate scope of this first asymmetric Brønsted acid catalysed reaction in aqueous media was examined (Table 4).
|
|
||||
|---|---|---|---|---|
| Entrya | R | Time/h | Yield (%)b | ee (%)c |
| a Reaction conditions: quinoline 2, Hantzsch dihydropyridine 4c (2.4 equiv.) and 2 mol% 1c, 1.4 M in brine at 50 °C. b Isolated yields after column chromatography. c Enantiomeric excess was determined by HPLC (Chiralcel OD-H). | ||||
| 1 | Phenyl | 30 | 81 | 92 |
| 2 | 3-Fluorophenyl | 40 | 60 | 90 |
| 3 | 3-Methylphenyl | 40 | 84 | 86 |
| 4 | 4-Ethylphenyl | 40 | 81 | 84 |
| 5 | 2-Naphthyl | 45 | 86 | 93 |
| 6 | 3-Bromophenyl | 40 | 90 | 86 |
| 7 | 4-(CF3)-Ph | 45 | 95 | 97 |
| 8 | 1,1′-Biphenyl-4-yl | 30 | 80 | 97 |
| 9 | 4-Methoxyphenyl | 40 | 85 | 91 |
| 10 | 2-Furyl | 40 | 80 | 87 |
| 11 | 4-Chlorophenyl | 40 | 85 | 96 |
| 12 | 4-Fluorophenyl | 40 | 81 | 93 |
| 13 | 3,4,5-Trimethoxyphenyl | 30 | 84 | 85 |
| 14 | 3-Methoxyphenyl | 40 | 93 | 83 |
In general, for the first time it was possible to isolate diverse aromatic and heteroaromatic tetrahydroquinolines with various substituents in good yields and with high enantioselectivities (Table 4). Compared to our previous results based on catalyst 1e in water-free benzene, these reactions conducted in aqueous NaCl solution were not only surprisingly consistent in terms of the enantioselectivities achieved but enable ecological and economical advantageous reaction processes.
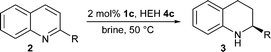
Subsequent to demonstrating for the first time that highly enantioselective Brønsted acid catalysis can be performed with water as the reaction medium, we decided to further demonstrate the application of this methodology in the transfer hydrogenation of cyclic imine 5. Using the optimized reaction conditions we were able to directly obtain chiral amine 6 with an enantiomeric excess of 90% (Scheme 1).18
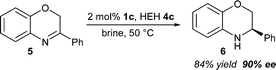 | ||
| Scheme 1 Brønsted acid catalysed reduction of benzoxazine 5. | ||
In order to gain more information on the mechanism and to determine the source of the proton in the enamine–imine tautomerisation step of the asymmetric transfer hydrogenation in aqueous media, we applied deuterated water as a reaction medium (Scheme 2). Formally, the reaction sequence involves a 1,4-hydride addition, protonation, and 1,2-hydride addition. In the following experiment the single (10) and double (11) deuterated tetrahydroquinoline were isolated almost exclusively.
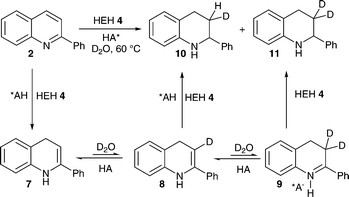 | ||
| Scheme 2 Brønsted acid catalysed deuteration in the reaction sequence 1,4-hydride addition, protonation and 1,2-hydride addition. | ||
Therefore, it can be assumed that in aqueous reaction media the dihydropyridine acts exclusively as a hydride source and not as the proton source. Further, the deuterium experiment shows that the enantioselective Brønsted acid catalysed transfer hydrogenation described not only occurs in the presence of water but that water plays an important role in the reaction procedure.
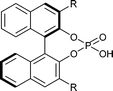
The good stereoinduction of the Brønsted acid 1c compared to 1e is exemplified by the molecular structures of the catalysts. For the first time we were able to determine the X-ray crystal structure of 1c (Fig. 2, left). The large isopropyl substituents act as a hydrophobic pocket in which the active centre of the catalyst is encased.19 In contrast to the open and water-accessible phenanthryl substituted phosphoric acid diester 1e (Fig. 2, right), this allows a stable contact ion pair to be formed which results in improved enantioselectivities and thereby enables the first asymmetric reaction procedure in aqueous reaction media.
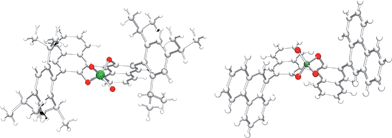 | ||
| Fig. 2 Molecular structures of Brønsted acids 1c (left) and 1e (right). | ||
In summary we have developed the first highly enantioselective Brønsted acid catalysed reaction using water as a reaction medium employing the principle of hydrophobic hydration. Further, it represents the first example in the field of organocatalysis of a non-covalent asymmetric induction conducted in a pure aqueous reaction solvent. This enantioselective Brønsted acid catalysed activation, previously considered impossible, provides, in the transfer hydrogenation described, an efficient route to 2-substituted tetrahydroquinolines or cyclic amines in good yields and with excellent enantioselectivities. The ecologically and economically advantageous reaction medium, water, further simplifies this already practical method and makes this reduction an attractive synthesis possibility for optically active tetrahydroquinolines and amines. Application on an industrial scale is also possible as the synthetic and biocatalytic recycling of dihydroypyridines in water is already feasible. Water as a reaction medium no longer excludes asymmetric Brønsted acid catalysis and it is only a question of time until further examples in this area are developed.
Acknowledgements
The authors acknowledge Evonik Degussa and the DFG (Priority Programme Organocatalysis) for financial support.Notes and references
- (a) D. C. Rideout and R. Breslow, J. Am. Chem. Soc., 1980, 102, 7816 CrossRef CAS; (b) R. Breslow, U. Maitra and D. C. Rideout, Tetrahedron Lett., 1983, 24, 1901 CrossRef CAS.
- For reviews see e.g.: (a) U. M. Lindström, Chem. Rev., 2002, 102, 2751 CrossRef; (b) S. Kobayashi and K. Manabe, Acc. Chem. Res., 2002, 35, 209 CrossRef CAS; (c) C.-J. Li, Chem. Rev., 2005, 105, 3095 CrossRef CAS; (d) A. Chanda and V. V. Fokin, Chem. Rev., 2009, 109, 725 CrossRef CAS. For reviews on asymmetric reactions in water see: (e) C. Pan and Z. Wang, Coord. Chem. Rev., 2008, 252, 736 CrossRef CAS; (f) J. Mlynarski and J. Paradowska, Chem. Soc. Rev., 2008, 37, 1502 RSC; (g) J. Paradowska, M. Stodulski and J. Mlynarski, Angew. Chem., Int. Ed., 2009, 48, 4288 CrossRef CAS; (h) M. Raj and V. K. Singh, Chem. Commun., 2009, 6687 RSC.
- (a) S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, Angew. Chem., 2005, 117, 3339 ( Angew. Chem., Int. Ed. , 2005 , 44 , 3275 ) CrossRef; (b) A. P. Brogan, T. J. Dickerson and K. D. Janda, Angew. Chem., Int. Ed., 2006, 45, 8100 CrossRef CAS; (c) Y. Hayashi, Angew. Chem., Int. Ed., 2006, 45, 8103 CrossRef CAS; (d) D. G. Blackmond, A. Armstrong, V. Coombe and A. Wells, Angew. Chem., 2007, 119, 3872 ( Angew. Chem., Int. Ed. , 2007 , 46 , 3798 ) CrossRef.
- Reviews: (a) M. Gruttadauria, F. Giacalone and R. Noto, Adv. Synth. Catal., 2009, 351, 33 CrossRef CAS. Selected examples: (b) Y. Hayashi, T. Sumiya, J. Takahashi, H. Gotoh, T. Urushima and M. Shoji, Angew. Chem., 2006, 118, 972 ( Angew. Chem., Int. Ed. , 2006 , 45 , 958 ) CrossRef; (c) N. Mase, Y. Nakai, N. Ohara, H. Yoda, K. Takabe, F. Tanaka and C. F. Barbas III, J. Am. Chem. Soc., 2006, 128, 734 CrossRef CAS; (d) S. Guizzetti, M. Benaglia, L. Raimondi and G. Celentano, Org. Lett., 2007, 9, 1247 CrossRef CAS; (e) V. Maya, M. Raj and V. K. Singh, Org. Lett., 2007, 9, 2593 CrossRef CAS; (f) J. Huang, X. Zhang and D. W. Armstrong, Angew. Chem., Int. Ed., 2007, 46, 9073 CrossRef CAS; (g) D.-Q. Xu, A.-B. Xia, S.-P. Luo, J. Tang, S. Zhang, J.-R. Jiang and Z.-Y. Xu, Angew. Chem., Int. Ed., 2009, 48, 3821 CrossRef.
- For nucleophilicity of primary and secondary amines, and amino acids in water, see: (a) F. Brotzel, Y. C. Chu and H. Mayr, J. Org. Chem., 2007, 72, 3679 CrossRef CAS; (b) F. Brotzel and H. Mayr, Org. Biomol. Chem., 2007, 5, 3814 RSC.
- (a) G. Lelais and D. W. C. MacMillan, Aldrichimica Acta, 2006, 39, 79 CAS; (b) S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471 CrossRef CAS.
- (a) S. Xu, Z. Wang, X. Zhang and K. Ding, Angew. Chem., Int. Ed., 2008, 47, 2840 CrossRef CAS; (b) S. Xu, Z. Wang, Y. Li, X. Zhang, H. Wang and K. Ding, Chem.–Eur. J., 2010, 16, 3021 CrossRef CAS; (c) M. Sickert, F. Abels, M. Lang, J. Sieler, C. Birkemeyer and C. Schneider, Chem.–Eur. J., 2010, 16, 2806 CrossRef CAS; (d) D. S. Giera, M. Sickert and C. Schneider, Org. Lett., 2008, 10, 4259 CrossRef CAS; (e) M. Sickert and C. Schneider, Angew. Chem., Int. Ed., 2008, 47, 3631 CrossRef CAS.
- (a) C. M. Kleiner and P. R. Schreiner, Chem. Commun., 2006, 4315 RSC; (b) Z. Zhang and P. R. Schreiner, Chem. Soc. Rev., 2009, 38, 1187 RSC.
- W. Blokzijl and J. B. F. N. Engberts, Angew. Chem., Int. Ed. Engl., 1993, 32, 1545 CrossRef.
- (a) W. Blokzijl, J. B. F. N. Engberts and M. J. Blandamer, J. Am. Chem. Soc., 1990, 112, 1197 CrossRef CAS; (b) R. Breslow, Acc. Chem. Res., 1991, 24, 159 CrossRef CAS; (c) W. Blokzijl, M. J. Blandamer and J. B. F. N. Engberts, J. Am. Chem. Soc., 1991, 113, 4241 CrossRef CAS; (d) J. B. F. N. Engberts, Pure Appl. Chem., 1995, 67, 823 CrossRef CAS; (e) S. Otto and J. B. F. N. Engberts, Org. Biomol. Chem., 2003, 1, 2809 RSC; (f) R. Breslow, Acc. Chem. Res., 2004, 37, 471 CrossRef CAS; (g) M. J. Blandamer, J. B. F. N. Engberts, P. T. Gleeson and J. C. R. Reis, Chem. Soc. Rev., 2005, 34, 440 RSC.
- C.-J. Li and L. Chen, Chem. Soc. Rev., 2006, 35, 68 RSC.
- R. C. Hudson and R. M. Daniel, Comp. Biochem. Physiol., 1993, 106B, 767 CAS.
- (a) N. Singh, S. J. Maniscalco and H. F. Fisher, J. Biol. Chem., 1993, 268, 21 CAS; (b) T. J. Stillman, P. J. Baker, K. L. Britton and D. W. Rice, J. Mol. Biol., 1993, 234, 1131 CrossRef CAS.
- (a) T. Akiyama, J. Itoh and K. Fuchibe, Adv. Synth. Catal., 2006, 348, 999 CrossRef CAS; (b) T. Akiyama, Chem. Rev., 2007, 107, 5744 CrossRef CAS; (c) A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713 CrossRef CAS; (d) M. Terada, Chem. Commun., 2008, 4097 RSC; (e) D. Kampen, C. M. Reisinger and B. List, Top. Curr. Chem., 2009, 291, 395.
- (a) S. G. Ouellet, A. M. Walji and D. W. C. Macmillan, Acc. Chem. Res., 2007, 40, 1327 CrossRef CAS; (b) S.-L. You, Chem.–Asian J., 2007, 2, 820 CrossRef CAS; (c) S. J. Connon, Org. Biomol. Chem., 2007, 5, 3407 RSC; (d) C. Wang, X. Wu and J. Xiao, Chem.–Asian J., 2008, 3, 1750 CrossRef CAS; (e) M. Rueping, E. Sugiono and F. R. Schoepke, Synlett, 2010, 852 CrossRef CAS.
- R. Breslow and C. J. Rizzo, J. Am. Chem. Soc., 1991, 113, 4340 CrossRef CAS.
- (a) M. Rueping, T. Theissmann and A. P. Antonchick, Synlett, 2006, 1071 CrossRef CAS; (b) M. Rueping, A. P. Antonchick and T. Theissmann, Angew. Chem., Int. Ed., 2006, 45, 3683 CrossRef; (c) M. Rueping, A. P. Antonchick and T. Theissmann, Angew. Chem., Int. Ed., 2006, 45, 6751 CrossRef CAS; (d) M. Rueping and A. P. Antonchick, Angew. Chem., Int. Ed., 2007, 46, 4562 CrossRef CAS; (e) M. Rueping and A. P. Antonchick, Angew. Chem., Int. Ed., 2008, 47, 5836 CrossRef CAS; (f) M. Rueping, T. Theissmann, S. Raja and J. W. Bats, Adv. Synth. Catal., 2008, 350, 1001 CrossRef CAS; (g) M. Rueping, F. Tato and F. R. Schoepke, Chem.–Eur. J., 2010, 16, 2688 CrossRef CAS; (h) M. Rueping, E. Sugiono, A. Steck and T. Theissmann, Adv. Synth. Catal., 2010, 352, 281 CrossRef CAS.
- In order to obtain the product and to use as little organic solvent as possible the aqueous solution was evaporated and directly filtered through silica.
- CPK views of the catalysts show clearly the steric demands and are shown in the ESI†.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, NMR and mass spectra and chromatograms. CCDC reference numbers 783865 and 783866. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00206b |
| This journal is © The Royal Society of Chemistry 2010 |