Ambient temperature anion-dependent spin state switching observed in “mostly low spin” heteroleptic iron(II) diimine complexes†
Zhaoping
Ni
,
Ashley M.
McDaniel
and
Matthew P.
Shores
*
Department of Chemistry, Colorado State University, Fort Collins, CO 80523-1872, USA. E-mail: shores@lamar.colostate.edu; Tel: (+1) 970-491-7235
First published on 26th August 2010
Abstract
We report the synthesis, characterization, and representative anion-binding studies on a series of heteroleptic FeII diimine complexes that have been designed to show anion-triggered spin state switching at ambient temperatures. Starting with [(H2bip)2FeBr2] (1), ligand substitution affords [(H2bip)2Fe(NN)]2+ complexes, where (NN) = pipi (2), bpy (3), and phen (4). In the solid state, the tetraphenylborate and bromide salts of the complexes display different thermally induced spin crossover properties, with spin transition temperatures above 395 K. The solid state magnetic properties depend on the FeII ligand field parameters and anion–cation interactions as well as solvent and packing effects. In dichloromethane solution, the weakly bound tetraphenylborate salts show spin state diminution, amplified chemical shift responses toward the diamagnetic 1H NMR spectral window, and visible colour changes upon titration of bromide anions (as nBu4N+ salts). Both 1H NMR- and electronic absorption-monitored titration studies unequivocally link anion binding to a high spin → low spin transition for the minority species in solution. Further, the complexes show pronounced air stability, in contrast to the homoleptic parent complex [Fe(H2bip)3]2+. Tuning the FeII ligand field by judicious ligand substitution toward “mostly low spin” species thus increases both the environmental stability and operating temperature of the complexes, and provides evidence that spin state switching may serve as a viable signalling event for chemical sensing in solution.
Introduction
Molecular species capable of switching between two states, such as spin crossover (SC) complexes, are increasingly pursued for potential applications in data storage and display devices.1,2 Reversible switching between low and high spin states (LS and HS, respectively) driven by small external perturbations such as temperature, pressure or light, stimulates dramatic physical responses in complex magnetism, polarisability and colour.3–7 Noting that the SC phenomenon is highly sensitive to small environmental changes, more recent attention has turned toward understanding and controlling chemical triggers, a necessary first step toward exploiting SC-based compounds in chemosensing schemes. This has been examined by several groups in the solid state, especially in metal–organic and metal–cyanide framework solids and polyhedra.8–12 Most notably, the presence or absence of neutral guest and/or solvate molecules in porous frameworks has been shown to induce spin state changes in the compounds via a steric “internal pressure” mechanism.13–16Since most (FeII) SC compounds are cationic,17 and anions play important roles in biological, industrial, and environmental processes,18–20 moving from neutral to anionic targets represents an attractive option for SC-based sensing architectures. Extensive efforts have been devoted to exploring the influence of anions and/or salts on SC properties in the solid state, and some useful structure–property correlations have been obtained.21,22 However, investigations of SC in the solid state are dependent on favourable crystallisations to establish robust correlations between host–anion interactions and spin state properties.21 Packing effects and the presence or absence of solvate molecules often thwart the clear observation of anion effects on SC properties and thus hinder the establishment of magnetostructural correlations.22 Probing guest-dependent spin state switching in solution offers an alternative method for assessing the effects of secondary non-covalent interactions on spin state properties.25,26 While the reporting of anion binding by diamagnetic coordination and organometallic complexes is an active area of research,23–27 efforts to achieve outer sphere anion-induced spin state changes in solution are scarce.28
We have focused our initial efforts on FeII complexes containing the ditopic H2bip ligand (Scheme 1), salts of which were recognised some thirty years ago as showing anion-dependent SC properties in the solid state.29 We have found that the solution spin state of the air-sensitive homoleptic complex [Fe(H2bip)3]2+ can be correlated to the extent of hydrogen-bonding interactions with charge balancing anions.30 Indeed, titration of anions capable of chelation by H2bip (such as bromide) results in the displacement of bulky tetraphenylborate anions and a concomitant diminution in magnetic susceptibility. This proof-of-concept spin state switching anion sensing phenomenon can be observed in CH2Cl2 solution at −40 °C.
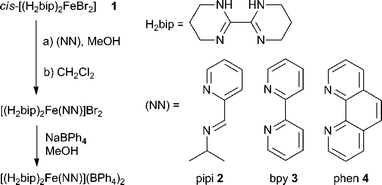 | ||
| Scheme 1 Preparation of heteroleptic iron(II) diimine complexes. | ||
Endeavouring to establish guest-dependent spin state switching as a viable chemosensing technique, it becomes clear that any applications would require the optimisation of this phenomenon toward higher operating temperatures and greater environmental robustness. We hypothesize that strengthening the ligand field encountered by the FeII ion may address both concerns simultaneously, since stronger fields stabilise the low spin state, which in turn increases SC temperatures and reduces complex lability.31 Of course, such optimisation requires synthetic tunability in the form of heteroleptic complexes, where some ligand(s) are responsible for anion binding while others adjust the ligand field into the near-SC regime. Here, we report the preparations of and initial anion titration studies on heteroleptic [(H2bip)2Fe(NN)]2+ complexes (NN = chelating N-donor ligand, Scheme 1). Importantly, we find that the incorporation of strong field ligands into the complex architecture gives rise to anion-dependent spin state switching at ambient temperatures with improved complex air stability, highlighting a potentially exploitable advantage in using “mostly LS” complexes in spin-switching chemosensing.
Results and discussion
Preparation and characterisation of heteroleptic diimine complex salts
Ligand field tunability requires developing a route for heteroleptic complexes, a challenge for labile first row transition metal ions. Whereas the homoleptic [Fe(H2bip)3]2+ complex forms readily in polar organic solvents, ligand scrambling would occur if multiple ligands were exposed to FeII simultaneously. Instead, we have discovered that sequential ligand substitution is possible by exploiting selective solubilities. The HS molecular compound cis-[(H2bip)2FeBr2] (1) can be prepared in good yield by the direct reaction of FeBr2 with stoichiometric H2bip in tetrahydrofuran (THF). Complex 1 is insoluble in THF, whereas any [Fe(H2bip)3]Br2 formed as a side product is readily soluble, and can be washed away from the product. The reaction of 1 with stoichiometric chelating (NN) ligands in methanol, followed by extraction into CH2Cl2, gives the desired heteroleptic [(H2bip)2Fe(NN)]Br2 complex salts in good yields (Scheme 1), where the ancillary (NN) ligands include 2-pyridinalisopropylimine (pipi, 2), 2,2′-bipyridine (bpy, 3), and 1,10-phenanthroline (phen, 4). Anion exchange reactions carried out with excess NaBPh4 in methanol afford the corresponding tetraphenylborate salts as analytically pure powdered precipitates. The tetraphenylborate salts of 2–4 are soluble in a variety of polar organic solvents; however, due to their propensity for ligand scrambling (vide infra), investigations of the tetraphenylborate salts were carried out in less polar dichloromethane solutions.The red-orange precursor complex 1 and the green, olive and indigo tetraphenylborate (BPh4−) salts of 2–4 (2·BPh4, 3·BPh4, 4·BPh4) have been analysed by single crystal X-ray crystallography. Crystallographic data and selected structural parameters are presented in Table S1.† The solid state structure of 1, acquired at room temperature, is shown in Fig. 1. The FeII ion is found in a N4Br2 distorted octahedral environment. The H2bip ligands show a cis orientation, presumably due to the higher steric hindrance of the α-hydrogen atoms in the alternative trans configuration.32 This results in a cis disposition of the bromide ligands, which makes their eventual substitution with a bidentate (NN) ligand more facile. The average Fe–N and Fe–Br bond distances are 2.132(9) and 2.762(2) Å, respectively, consistent with a HS FeII ion. The measured octahedral (Σ)33,34 and trigonal (Θ)35 distortion parameters for 1 (Table S1†) are consistent with other structurally characterised HS FeII complexes. The complexes pack together such that the bound bromide anions are chelated by the N–H moieties of H2bip ligands on neighbouring complexes, in which the hydrogen bonding interactions of N–H⋯Br range from 3.351(9) to 3.400(9) Å (Fig. S1†). The shortest intermolecular Fe⋯Fe distance is 8.075(3) Å.
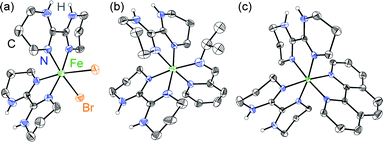 | ||
| Fig. 1 Crystal structures of (a) 1 and the cations in (b) 2·BPh4 and (c) 4·BPh4, rendered with 40% ellipsoids. Only H atoms bound to N atoms are retained for clarity. | ||
The crystal structures of 2·BPh4–4·BPh4 have been investigated at 100 K, and the cationic complexes in 2·BPh4 and 4·BPh4 are shown in Fig. 1. We note that ligand disorder in bpy-containing 3·BPh4 precludes quantitative structural comparisons with the pipi- and phen-containing compounds 2·BPh4 and 4·BPh4 but nevertheless provides support for the expected structure (see ESI†). The anticipated FeII coordination by two H2bip ligands and one ancillary (NN) ligand is observed, Fe–N distances are as expected for LS FeII ions, and Σ and Θ distortion parameters (Table S1†) indicate a more regular octahedral environment typical for FeII in the LS state.36 Minimal hydrogen bonding interactions between the cation and BPh4− anions are observed—the shortest NH⋯BPh4 is 3.197(3) Å in 4·BPh4—and in no case do the H2bip ligands chelate a BPh4− species. The cations are separated by the bulky BPh4− anions and the shortest intermetallic distances are 12.048(1) and 10.985(1) Å in 2·BPh4 and 4·BPh4, respectively.
The ancillary (NN) ligands were selected for their expected augmentation of the heteroleptic complex ligand field according to the following expressions:
| Δ[(H2bip)2Fe(NN)] = (2Δ[Fe(H2bip)3] + Δ[Fe(NN)3])/3 | (1) |
| Δ[Fe(L)3] = cΔ[Ni(L)3] (1 ≤ c ≤ 1.1) | (2) |
Notwithstanding, ambient temperature solid state magnetic susceptibility measurements for both BPh4− and Br− salts of complexes 2–4 display χMT values in the range 0.15–0.64 cm3 K mol−1 (Fig. 2), indicating that any spin equilibrium favours the diamagnetic LS state at room temperature. Negligible increases in χMT values are observed for 3·Br, 3·BPh4 and 4·Br up to 395 K, while 2·Br, 2·BPh4 and 4·BPh4 show gradual and incomplete spin crossover. The largest χMT value at 395 K is 1.47 cm3 K mol−1 for 4·BPh4, indicating that all the solid state spin crossover transition temperatures (T1/2) are larger than 395 K. Not surprisingly, and consistent with numerous studies of FeII complexes in the solid state, the observed spin states depend on not only the ligand field, but also packing effects driven by the charge-balancing anions and any solvate molecules.39,40 The latter can be confirmed by the different χMT curves for 4·BPh4 with and without included CH2Cl2 solvent (Fig. S5†). The former is best illustrated by the pipi-containing complex 2: although infrared spectra show more significant hydrogen bonding for the Br− salt compared to the BPh4− salt (via ΔνN–H), the solid state χMT value for 2·Br is slightly larger than for 2·BPh4; the opposite would be expected if spin states were determined only by ligand field and anion–cation interactions.
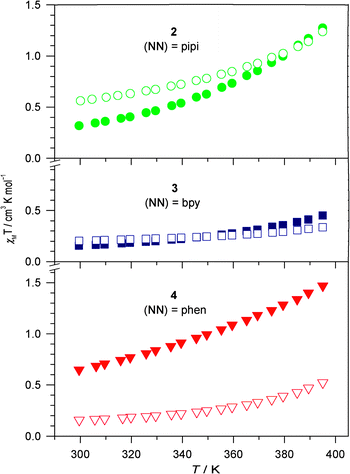 | ||
| Fig. 2 Temperature dependence (cool down) of the solid state magnetic susceptibilities for 2–4 with Br− (open) and BPh4− (closed) charge balancing anions. The samples were desolvated by heating to 395 K. | ||
Whereas the solid state magnetic properties result from multiple contributions (e.g. ligand field, anion–cation interactions, crystal packing, presence/absence of solvate molecules), the solution magnetic properties (measured by the Evans method40–43) appear to track more consistently with the identity of the charge balancing anion. For all three heteroleptic complexes, 1H NMR spectra obtained in CD2Cl2 solution at room temperature indicate significant differences between the Br− and BPh4− salts in that the former show diamagnetic 1H NMR spectra,41 while the latter display paramagnetically shifted signals (Fig. S6, 9–10†), with χMT values of 0.69, 0.43 and 0.54 cm3 K mol−1 at 296 K for 2·BPh4–4·BPh4, respectively.42 These results suggest that the magnetic data in solution are more regular than those in the solid state and the hydrogen bonding interactions favour the LS state. The magnetic data indicate that even the BPh4− salts of complexes 2–4 are “mostly LS” compounds at room temperature.
The nature of solution SC properties for 2·BPh4–4·BPh4 is confirmed from variable temperature 1H NMR spectra collected between 193 and 308 K (Fig. 3). All the χMT values gradually diminish with decreasing temperature. We notice a concomitant decline in the chemical shift values of the α proton on the (NN) ligands; this upfield tracking into the “normal” 1H NMR window is consistent with a shift to the pure LS state upon decreasing the temperature. The susceptibility data can be fit to a HS–LS equilibrium (modified from literature precedent43), and give reasonable thermodynamic parameters for SC behavior in solution3,31,44,45 so long as we include a small amount (1–2%) of [Fe(H2bip)3]2+ impurity (vide infra). See the ESI for details of the fitting procedure.† In sum, the solution behavior shows gradual and incomplete spin crossover in solution for the three tetraphenylborate complex salts.
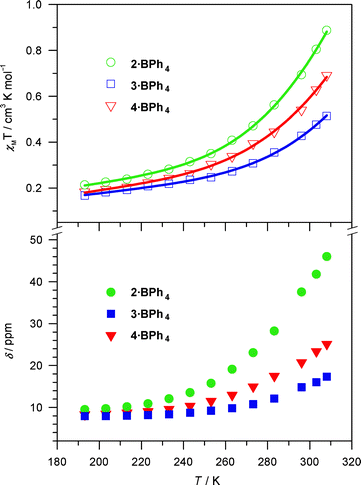 | ||
| Fig. 3 Temperature dependence of the solution magnetic susceptibilities (top) and chemical shifts of α protons on the (NN) ligand (bottom) for 2·BPh4–4·BPh4 in CD2Cl2. Only the chemical shift for the imine H is shown for 2·BPh4. Lines represent fits of the data to a HS–LS equilibrium. See text and ESI for details of the fitting procedure.† | ||
Tuning the strength of the ancillary (NN) ligands, the dissolved BPh4− salts display different colours due to ligand-induced charge transfer band shifts: green for 2·BPh4, olive for 3·BPh4, and indigo for 4·BPh4. Importantly, the spectra of 2–4 also vary as a function of anion. When the charge balancing anion is changed from BPh4− to Br−, the absorption bands show obvious red shifts (Δλmax = 25 nm for 2·Br (green), 31 nm for 3·Br (olive) and 26 nm for 4·Br (teal)) consistent with significant N–H⋯Br− interactions.
Anion binding investigations
Because we are primarily interested in the effect that representative cation–anion interactions have on spin state switching for these “mostly LS” complexes rather than the optimisation of host:guest binding, we have restricted initial anion binding studies to Br−, since the previously reported [Fe(H2bip)3]2+ complex is moderately selective for Br− over Cl−, I−, NO3− and ClO4−.32 Here, we observe small but significant drops in solution χMT values when 2·BPh4–4·BPh4 are interrogated with Br− (as nBu4NBr) at room temperature. Indeed, the χMT value for the pipi complex 2·BPh4 decreases almost linearly from 0.69 to 0.31 cm3 K mol−1 when titrating zero to 2.5 equivalents of Bu4NBr (Fig. 4a). More gradual changes are found for 3·BPh4 (from 0.43 to 0.24 cm3 K mol−1) and 4·BPh4 (from 0.54 to 0.36 cm3 K mol−1) after adding 2.5 equivalents of Br−. These changes are more subtle than what was observed for the analogous titration performed on the homoleptic [Fe(H2bip)3]2+ complex, even accounting for the change in temperature.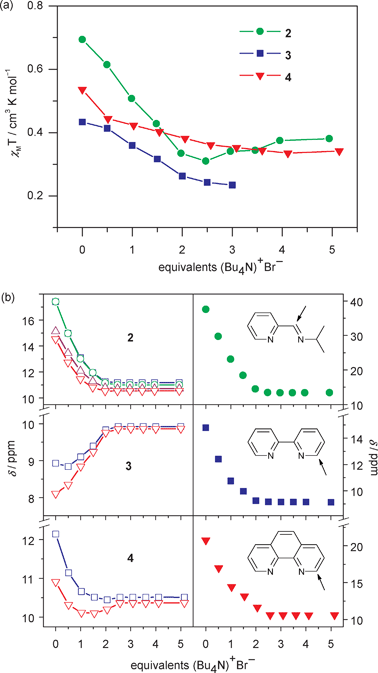 | ||
| Fig. 4 Changes in ambient temperature (296 K) (a) magnetic susceptibility and (b) chemical shifts of N–H protons on the H2bip ligand (left) and α protons on the (NN) ligand (right) upon the addition of nBu4NBr to CD2Cl2 solutions of 2·BPh4–4·BPh4. The χMT value is too small to be observed for 3·BPh4 after adding three eq. of Br−. Only the chemical shift for the imine H is shown for 2·BPh4. Lines are guides to the eye. | ||
While titration-induced solution magnetic susceptibility decreases are modest, investigation of the chemical shift data reveals more substantial changes. In all cases, chemical shifts (δ) of the (NN) ligand protons in 2–4 track upfield into the “normal” 1H NMR window as up to 2.5 equivalents of Br− are titrated (Fig. S11†). In contrast to the limited change in δ for organic anion receptors or purely diamagnetic metallic coordination complex hosts,46–49 a large upfield shift (−24.1 ppm) is observed for one of the α (imine) protons on the pipi ligand in 2·BPh4 (Fig. 4b). Complexes 3·BPh4 and 4·BPh4 also show obvious upfield shifts of −5.6 and −10.1 ppm, respectively. Therefore, subtle spin state changing can result in an amplified chemical shift response in these “mostly LS” complexes. Meanwhile, the H2bip N–H resonances in 2·BPh4 track with the (NN) C–H chemical shift changes described above, albeit with smaller magnitudes. To a lesser extent, this is also observed for the N–H signals in 4·BPh4. However, the N–H signals in 3·BPh4 actually show downfield shifts upon addition of up to 2.5 equivalents of Bu4NBr.
The observed anion-triggered changes in δ can be understood as the result of competing interactions. Downfield shifting upon anion binding is common in organic or diamagnetic coordination compound receptors,46–49 where H-bonding interactions with the anion reduce electron density on the donor atom, thus deshielding the proton and provoking a positive Δδ for the N–H signal.50 Because bpy-containing 3·BPh4 has a smaller magnetic moment than pipi-containing 2·BPh4 at room temperature, it is not surprising that this effect dominates. Meanwhile, upfield shifts exhibited by the (NN) ligand protons are linked solely to SC properties.44,45 Since the conversion in solution between HS and LS states is much faster than the time scale of the NMR experiment, only the average δ for the HS and LS forms is measured. For FeII complexes, paramagnetic shifting of δ depends only on the HS : LS ratio. When the magnetic moment is reduced (i.e. when Br− is added to the system to form a diamagnetic LS species), the δ values track toward the “normal” proton window, resulting in upfield shifts. Thus, changes in the (NN) ligand δ values offer a robust probe of anion binding and concomitant spin state changes in solution, even in the absence of large susceptibility changes.
We note that at first glance, ligand dissociation is a potential complicating factor in these analyses, especially for the minor component HS FeII species.51 The 1H NMR spectra of freshly prepared CD2Cl2 solutions of analytically pure complex salts of 2·BPh4–4·BPh4 show a small amount (<3%) of [Fe(H2bip)3](BPh4)2, perhaps due to ligand rearrangement even in the relatively nonpolar solvent.52 Weak signals not attributable to 2–4 are observed in the “normal” proton window, but none correspond to [Fe(NN)3]2+ species. The most simple conjecture is that an equimolar amount of [(H2bip)Fe(NN)2]2+ (relative to [Fe(H2bip)3]2+) is present in solution. These heteroleptic impurities are expected to be LS based on ligand field considerations, and while they and any [Fe(H2bip)3]2+ may compete for anions with 2–4, the spin states of [(H2bip)Fe(NN)2]2+ are unlikely to change upon binding with anions.
No other obvious spectral changes occur between fresh solutions and those left standing for 140 min, a time period longer than required to carry out the titration experiments. However, addition of more than 2.5 eq. Br− to fresh solutions of the BPh4− salts gives NMR signals attributable to re-formation of precursor 1, which increases solution χMT values slightly; this is most obvious for the pipi-containing 2·BPh4 (Fig. 4a). Based on these observations, we concede that impurities and/or ligand rearrangement products may obscure magnetic susceptibility information, especially for the “mostly LS” complexes studied here.
Notwithstanding, we also observe that δ values for 2·BPh4–4·BPh4 change less than 0.2 ppm after adding 0.5 eq. of paramagnetic [Fe(H2bip)3](BPh4)2 (Fig. S12–14†). The ratiometric nature of the spin-switching response means that chemical shift information for the FeII host complexes is minimally affected by impurities, paramagnetic or otherwise, so long as they cannot interact chemically with the complexes. Whereas ligand dissociation may reduce slightly the number of spin-switching-capable species in solution, and will generate competitors for anions, it cannot affect the HS : LS ratio of the sensing complex. In sum, the chemical shift data demonstrate the direct spin-state reducing effect of anion interactions with the minority HS FeII complexes in solution.
Electronic absorption-monitored titrations carried out on salts of complexes 2–4 in CH2Cl2 offer an alternative probe of anion-triggered spin state switching. Titrations of 2·BPh4–4·BPh4 with Br− in dichloromethane do significantly shift the absorption maxima and change the absorption intensities after the addition of two equivalents of nBu4NBr (Fig. 5). This is most obvious for the phen-containing complex 4, which undergoes an indigo-to-teal color change monitored easily by the naked eye.
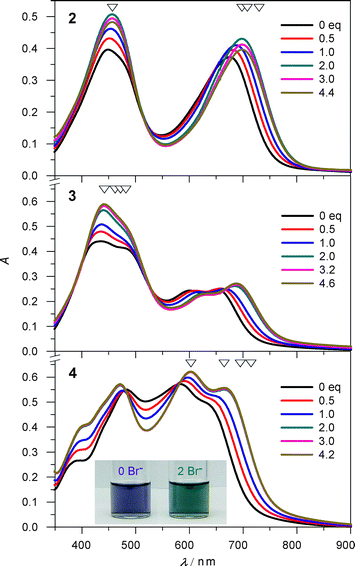 | ||
| Fig. 5 Changes in electronic absorption for 2·BPh4–4·BPh4 upon addition of nBu4NBr in CH2Cl2. Inset: colour changes of 4·BPh4 (1.1 × 10−4 M) upon addition of 0 (indigo) and 2 (teal) equivalents of nBu4NBr. Four wavelengths (open triangles) were selected for fitting the binding constants. | ||
Efforts to quantify host–guest interactions are based on the equilibria presented in Scheme 2. The most straightforward model (2K), where H2bip ligands provide the only anion-binding sites, leads to a neutral complex with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 host:guest stoichiometry. For the 2K model, binding curves can be analysed by the following:
:2 host:guest stoichiometry. For the 2K model, binding curves can be analysed by the following:
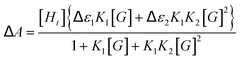 | (3) |
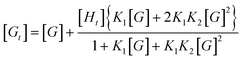 | (4) |
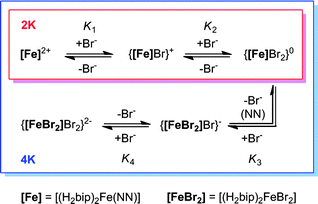 | ||
| Scheme 2 Models for anion binding studies carried out on 2·BPh4–4·BPh4. | ||
| 2·BPh4 | 3·BPh4 | 4·BPh4 | ||
|---|---|---|---|---|
| 2K: | log K1 | 6.3 | 6.2 | |
| log K2 | 4.9 | 5.6 | ||
| 4K: | log K1 | 6.0 | 6.7 | 6.6 |
| log K2 | 5.0 | 4.7 | 5.4 | |
| log K3 | 2.7 | 0.7 | 0.2 | |
| log K4 | 2.4 | 3.5 | 2.7 | |
| log K1 + log K2 | 11.0 | 11.4 | 12.0 | |
| log K3 + log K4 | 5.1 | 4.2 | 2.9 |
Unfortunately, attempts to obtain reasonable fits of the titration data to this simple model are successful only for complexes 3·BPh4 and 4·BPh4. However, reasonable fits for all three heteroleptic species 2–4 can be obtained if additional reactions are considered (4K model, Scheme 2). Here, two steps are collected in the third equilibrium constant: incorporation of bromide and loss of a chelating ligand, the latter to open up two potential anion-binding sites on the FeII ion (to form {[(H2bip)2FeBr2]⊂Br}). The binding curves are analysed by the following equations and the derived association constants are presented in Table 1:
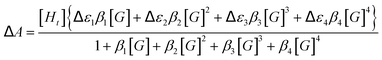 | (5) |
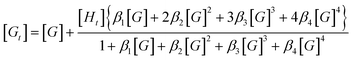 | (6) |
Comparison of the 2K and 4K models applied to the behaviours of 3·BPh4 and 4·BPh4 show qualitatively good agreement for the stepwise binding constants K1 and K2 (Table 1). The anion-binding abilities for the heteroleptic complexes are reflected in the sums of log K1 and log K2, while the degree of ligand dissociation is indicated by the sum of log K3 and log K4. We note that even the 4K model is simplistic, and ignores probable ligand dissociation in the host solution. Nevertheless, these titration studies show relatively strong binding of bromide to the heteroleptic complexes in dichloromethane solution, consistent with our previous results for [Fe(H2bip)3](BPh4)2.30
Anion binding studies with 2.5 equivalents of different anions Bu4NX (X = Cl, Br, I, NO3, ClO4) were performed for 4·BPh4 in dichloromethane. Judging by the chemical shifts of the α protons on the phen ligand as well as electronic absorption changes (Fig. S17†), the binding abilities track in the order Br− > Cl− > I− > NO3− > ClO4−, which is consistent with the behaviour of the homoleptic [Fe(H2bip)3]2+ complex.30 The distinct colour changes from indigo to teal are easily observed by the naked eye for the host solution interrogated with Cl− and Br−, somewhat weaker shifts are observed with I− and NO3−, while only a faint colour change occurs with the ClO4− anion. The combined spectroscopic studies argue compellingly for a model where the strength of hydrogen bonding interactions is a key driver in the spin state properties of the heteroleptic complexes.
We note that the indigo-to-teal colour change arising from the Br− titration of phen-containing 4·BPh4 is persistent in oxidising environments. Dichloromethane solutions of “mostly LS” 4·BPh4 exposed to air for several hours continue to display the anion-response function; in contrast, ambient temperature solutions of the HS parent complex [Fe(H2bip)3]2+ oxidise almost immediately upon exposure to air.
Conclusions
We have prepared a family of “mostly LS” heteroleptic FeII complexes that display anion-dependent spin state switching properties at room temperature. We find that ligand field considerations can be effective in narrowing the choice of ancillary ligands that will move spin transition properties to higher temperatures. However, while the bpy-containing complex 3 exerts a ligand field intermediate between 2 and 4, it shows the largest propensity for a LS configuration. Clearly, other more subtle considerations remain to be uncovered. Regardless, a benefit to using LS-leaning species is enhanced chemical stability: for the phen-containing complex 4, the colorimetric anion response phenomenon persists in air for several hours at the minimum.In contrast to diamagnetic hosts, we find that subtle spin state changing can provide amplified chemical shift responses. The magnetically driven chemical shift changes also isolate anion binding to the chemosensing species in the presence of small impurities, which may be useful in the analysis of complex mixtures. Further, the unambiguous observation of visible spectrum changes portends the development of a new class of naked-eye detection materials for anions.54–56 Certainly, achieving that requires translating the spin-switching sensing event to more competitive solvents, where anion binding is expected to be significantly weaker. On the other hand, the success noted here is amenable to improvement simply by incorporating more sophisticated anion-binding ligands into the sensing architecture. Efforts to prepare chemosensing architectures with high selectivity and environmental practicality are underway.
Acknowledgements
This research was supported by Colorado State University and the ACS Petroleum Research Fund (44691-G3). We thank Mr. C. Andrews for collecting some of the magnetometry data for 1, Dr W. Tong for the use of spectral fitting software, Dr C. D. Rithner for assistance with paramagnetic 1H NMR data collection and interpretation, and Prof. C. M. Elliott for helpful discussions.Notes and references
- O. Kahn and C. J. Martinez, Science, 1998, 279, 44–48 CrossRef CAS.
- Spin Crossover in Transition Metal Compounds I–III, eds. P. Gütlich and H. A. Goodwin, Springer, Berlin, 2004 Search PubMed.
- P. Gütlich, Y. Garcia and H. A. Goodwin, Chem. Soc. Rev., 2000, 29, 419–427 RSC.
- M. A. Halcrow, Chem. Soc. Rev., 2008, 37, 278–289 RSC.
- H.-J. Krüger, Coord. Chem. Rev., 2009, 253, 2450–2459 CrossRef.
- I. Šalitroš, N. T. Madhu, R. Boča, J. Pavlik and M. Ruben, Monatsh. Chem., 2009, 140, 695–733 CrossRef CAS.
- B. Weber, Coord. Chem. Rev., 2009, 253, 2432–2449 CrossRef CAS.
- T. Glaser, Angew. Chem., Int. Ed., 2003, 42, 5668–5670 CrossRef CAS.
- M. Nihei, L. Han and H. Oshio, J. Am. Chem. Soc., 2007, 129, 5312–5313 CrossRef CAS.
- G. Agustí, R. Ohtani, K. Yoneda, A. B. Gaspar, M. Ohba, J. F. Sánchez-Royo, M. C. Muñoz, S. Kitagawa and J. A. Real, Angew. Chem., Int. Ed., 2009, 48, 8944–8947 CrossRef CAS.
- M. Ohba, K. Yoneda, G. Agustí, M. C. Muñoz, A. B. Gaspar, J. A. Real, M. Yamasaki, H. Ando, Y. Nakao, S. Sakaki and S. Kitagawa, Angew. Chem., Int. Ed., 2009, 48, 4767–4771 CrossRef CAS.
- K. Ono, M. Yoshizawa, M. Akita, T. Kato, Y. Tsunobuchi, S.-i. Ohkoshi and M. Fujita, J. Am. Chem. Soc., 2009, 131, 2782–2783 CrossRef CAS.
- G. J. Halder, C. J. Kepert, B. Moubaraki, K. S. Murray and J. D. Cashion, Science, 2002, 298, 1762–1765 CrossRef CAS.
- G. J. Halder, K. W. Chapman, S. M. Neville, B. Moubaraki, K. S. Murray, J.-F. Létard and C. J. Kepert, J. Am. Chem. Soc., 2008, 130, 17552–17562 CrossRef CAS.
- S. M. Neville, G. J. Halder, K. W. Chapman, M. B. Duriska, B. Moubaraki, K. S. Murray and C. J. Kepert, J. Am. Chem. Soc., 2009, 131, 12106–12108 CrossRef CAS.
- P. D. Southon, L. Liu, E. A. Fellows, D. J. Price, G. J. Halder, K. W. Chapman, B. Moubaraki, K. S. Murray, J.-F. Létard and C. J. Kepert, J. Am. Chem. Soc., 2009, 131, 10998–11009 CrossRef CAS.
- M. A. Halcrow, Polyhedron, 2007, 26, 3523–3576 CrossRef CAS.
- F. Davis, S. D. Collyer and S. P. J. Higson, Top. Curr. Chem., 2005, 255, 97–124 CAS.
- J. L. Sessler, P. A. Gale and W.-S. Cho, Anion Receptor Chemistry, The Royal Society of Chemistry, Cambridge, U.K., 2006 Search PubMed.
- G. W. Bates and P. A. Gale, Struct. Bonding, 2008, 129, 1–44 CAS.
- M. Hostettler, K. W. Törnroos, D. Chernyshov, B. Vangdal and H.-B. Bürgi, Angew. Chem., Int. Ed., 2004, 43, 4589–4594 CrossRef CAS.
- M. Yamada, H. Hagiwara, H. Torigoe, N. Matsumoto, M. Kojima, F. Dahan, J.-P. Tuchagues, N. Re and S. Iijima, Chem. Eur. J., 2006, 12, 4536–4549 CrossRef CAS.
- P. D. Beer and S. R. Bayly, Top. Curr. Chem., 2005, 255, 125–162 CAS.
- C. R. Rice, Coord. Chem. Rev., 2006, 250, 3190–3199 CrossRef CAS.
- S. R. Bayly and P. D. Beer, Struct. Bonding, 2008, 129, 45–94 CAS.
- J. Pérez and L. Riera, Chem. Soc. Rev., 2008, 37, 2658–2667 RSC.
- J. W. Steed, Chem. Soc. Rev., 2009, 38, 506–519 RSC.
- To clarify, we wish to differentiate outer sphere “sensing” from the direct coordination of an anion to a SC center, as observed in [(phen)2Fe(NCS)2]. References: (a) E. Koenig and K. Madeja, Inorg. Chem., 1967, 6(1), 48–55 CrossRef and references therein; (b) E. König and K. Madeja, Chem. Commun., 1966, 61–62 RSC.
- M. G. Burnett, V. McKee and S. M. Nelson, J. Chem. Soc., Dalton Trans., 1981, 1492–1497 RSC.
- Z. Ni and M. P. Shores, J. Am. Chem. Soc., 2009, 131, 32–33 CrossRef CAS.
- P. Gütlich, A. Hauser and H. Spiering, Angew. Chem., Int. Ed. Engl., 1994, 33, 2024–2054 CrossRef.
- E. D. McKenzie, Coord. Chem. Rev., 1971, 6, 187–216 CrossRef CAS.
- M. G. B. Drew, C. J. Harding, V. McKee, G. G. Morgan and J. Nelson, J. Chem. Soc., Chem. Commun., 1995, 1035–1038 RSC.
- P. Guionneau, M. Marchivie, G. Bravic, J.-F. Létard and D. Chasseau, J. Mater. Chem., 2002, 12, 2546–2551 RSC.
- M. Marchivie, P. Guionneau, J.-F. Létard and D. Chasseau, Acta Crystallogr., Sect. B: Struct. Sci., 2005, 61, 25–28 CrossRef.
- G. Dupouy, M. Marchivie, S. Triki, J. Sala-Pala, J.-Y. Salaün, C. J. Gómez-García and P. Guionneau, Inorg. Chem., 2008, 47, 8921–8931 CrossRef CAS.
- M. A. Robinson, J. D. Curry and D. H. Busch, Inorg. Chem., 1963, 2, 1178–1181 CrossRef CAS.
- H. A. Goodwin, Top. Curr. Chem., 2004, 233, 59–90 CAS.
- D. L. Reger, J. R. Gardinier, M. D. Smith, A. M. Shahin, G. J. Long, L. Rebbouh and F. Grandjean, Inorg. Chem., 2005, 44, 1852–1866 CrossRef CAS.
- D. L. Reger, J. R. Gardinier, J. D. Elgin, M. D. Smith, D. Hautot, G. J. Long and F. Grandjean, Inorg. Chem., 2006, 45, 8862–8875 CrossRef CAS.
- They are too small to be observed for all the Br salts due to the overlap of the TMS peaks.
- Accounting for the presence of impurities (based on the NMR peak integration), the HS fractions for 2·BPh4–4·BPh4 at 296 K are 19%, 10% and 14%, respectively, assuming the limiting LS and HS magnetic susceptibilities of 0 and 3.38 cm3 K mol−1, respectively. Reference: J. C. Yoder, J. P. Roth, E. M. Gueeenhoven, A. S. Larsen and J. M. Mayer, J. Am. Chem. Soc., 2003, 125, 2629–2640 Search PubMed.
- J. F. Berry, F. A. Cotton, T. Lu and C. A. Murillo, Inorg. Chem., 2003, 42, 4425–4430 CrossRef CAS.
- K. P. Bryliakov, E. A. Duban and E. P. Talsi, Eur. J. Inorg. Chem., 2005, 72–76 CrossRef CAS.
- J. England, G. J. P. Britovsek, N. Rabadia and A. J. P. White, Inorg. Chem., 2007, 46, 3752–3767 CrossRef CAS.
- F. P. Schmidtchen and M. Berger, Chem. Rev., 1997, 97, 1609–1646 CrossRef CAS.
- C. Caltagirone and P. A. Gale, Chem. Soc. Rev., 2009, 38, 520–563 RSC.
- P. A. Gale, S. E. García-Garrido and J. Garric, Chem. Soc. Rev., 2008, 37, 151–190 RSC.
- S. Kubik, Chem. Soc. Rev., 2009, 38, 585–605 RSC.
- D. Makuc, Triyanti, M. Albrecht, J. Plavec, K. Rissanen, A. Valkonen and C. A. Schalley, Eur. J. Org. Chem., 2009, 4854–4866 CrossRef CAS.
- H. Taube, Chem. Rev., 1952, 50, 69–126 CrossRef CAS.
- Based on the relative intergration value, the contents of [Fe(H2bip)3](BPh4)2 are 1.5, 2.6 and 2.0% for 2·BPh4–4·BPh4, respectively. We do not observe [Fe(NN)3]2+ and free NN ligands in the 1H NMR spectra even after one day.
- K. A. Connors, Binding Constants: The Measurement of Molecular Complex Stability, John Wiley and Sons, New York, 1987 Search PubMed.
- C. Suksai and T. Tuntulani, Chem. Soc. Rev., 2003, 32, 192–202 RSC.
- R. Martínez-Máñez and F. Sancenón, Chem. Rev., 2003, 103, 4419–4476 CrossRef CAS.
- T. Gunnlaugsson, M. Glynn, G. M. Tocci, P. E. Kruger and F. M. Pfeffer, Coord. Chem. Rev., 2006, 250, 3094–3117 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and full spectroscopic characterisations. CCDC reference numbers 768562–768565. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00303d |
| This journal is © The Royal Society of Chemistry 2010 |