Photoinduced N2 loss as a route to long-lived organometallic alkane complexes: A time-resolved IR and NMR study†
James A.
Calladine
a,
Olga
Torres
b,
Mitch
Anstey
c,
Graham E.
Ball
*d,
Robert G.
Bergman
*c,
John
Curley
c,
Simon B.
Duckett
*b,
Michael W.
George
*a,
Amy I.
Gilson
c,
Douglas J.
Lawes
d,
Robin N.
Perutz
*b,
Xue-Zhong
Sun
a and
K. Peter C.
Vollhardt
*c
aSchool of Chemistry, University of Nottingham, University Park, Nottingham, UK NG7 2RD. E-mail: mike.george@nottingham.ac.uk
bDepartment of Chemistry, University of York, Heslington, York, UK YO10 5DD. E-mail: rnp1@york.ac.uk
cDepartment of Chemistry, University of California, Berkeley, California, 94270, USA. E-mail: rbergman@berkeley.edu; kpcv@berkeley.edu
dSchool of Chemistry, University of New South Wales, UNSW Sydney, NSW, 2052, Australia. E-mail: g.ball@unsw.edu.au
First published on 27th August 2010
Abstract
Photolysis of CpRe(CO)2(N2) in cyclopentane or 2,2-dimethylbutane with a UV lamp via a quartz fibre inserted into the NMR probe allows generation of CpRe(CO)2(cyclopentane) and CpRe(CO)2(2,2-dimethylbutane). The latter is observed in three isomeric forms according to the site of co-ordination to the rhenium. The major isomer, CpRe(CO)2(2,2-dimethylbutane-η2-C1,H1), exhibits a 1H NMR resonance for the co-ordinated hydrogen at δ = −2.19 with 1JC–H = 118 Hz. The photochemistry of Cp‡Re(CO)2(N2) (Cp‡ = η5-1,2-C5H3(tBu)2) in alkane solution is also reported. Two new organometallic alkane complexes, Cp‡Re(CO)2(alkane) (alkane = cyclopentane, n-heptane) have been characterized by IR spectroscopy following irradiation of Cp‡Re(CO)2(N2) and their rate constants for reaction with CO have been determined. The reaction with cyclopentane has also been studied by NMR spectroscopy at 190 K with in situ laser irradiation at 355 nm. Cp‡Re(CO)2(c-C5H10) is shown to exhibit the characteristic features of an alkane complex in the NMR spectrum, viz. a large isotopic shift of the 1H resonance at δ = −2.44 upon partial deuteration of the alkane (Δδ = 1.77 ppm), a large 1JC–H (114 Hz) and a large negative 13C chemical shift (δ = −33.8). We find no evidence for CO loss or agostic interactions of the t-butyl groups under these conditions. Cp‡Re(CO)2(alkane) has a slightly shorter lifetime (ca. 5x) than CpRe(CO)2(alkane) for a given alkane. Photolysis of CpRe(CO)2(N2) to form the organometallic alkane complex occurs with a much higher yield than for CpRe(CO)3. Efficient photo-ejection of N2 from Cp‡Re(CO)2(N2) is observed upon either 266 or 355 nm laser irradiation. A dinitrogen precursor allows for the use of longer wavelength irradiation and the generation of a higher concentration of the alkane complex following each laser pulse.
Introduction
Transition-metal σ-complexes are formed when the σ-bond of a saturated molecule of the type E–H (where E = C, H, B or Si) is donated to a transition-metal centre, in a 3-centre-2-electron interaction.1 The four principal classes of σ-complexes are, dihydrogen, silane, borane and alkane. Alkane complexes are of fundamental interest because of their key role as intermediates in C–H activation processes, whether involving oxidative addition or σ-CAM mechanisms.2–9 Organometallic alkane complexes were originally observed and characterized following irradiation of metal carbonyl precursors such as M(CO)6 (M = Cr, Mo or W) and Fe(CO)5 trapped in methane matrices at very low temperature (12 K).10,11 In solution at room temperature, Cr(CO)5(c-C6H12) was found to be formed within 50 ns following photolysis of Cr(CO)6 in cyclohexane solution and observed to decay on the microsecond timescale.12 This reflects the high reactivity of such systems, and fast time-resolved infrared (TRIR) spectroscopy has proven to be a powerful tool for monitoring both the formation and reactivity of organometallic alkane complexes.13–18 For example, it has played a key role in monitoring the C–H activation reactions which convert Rh-alkane species to Rh-alkyl hydrides for complexes such as Cp′Rh(CO)2 (Cp′ = η5-C5H5 or η5-C5Me5)19,20 and Tp′Rh(CO)2(alkane) (Tp′ = Tp3,5-Me or Tp4-tBu-3,5-Me; Tp = tris-pyrazolyl borate).21,22 The reactivity of the related alkane complexes (η5-C5R5)M(CO)2(alkane) (R = H, Me, Et or Ph; M = Mn or Re),23,24 M(CO)5(alkane) (M = Cr, Mo or W)25 and (η6-C6R6)Cr(CO)2(alkane) (R = H, Me or Et)26 proved to be affected by both the nature of the aryl substituents and the choice of alkane used.A systematic study of related metal carbonyl alkane complexes by TRIR spectroscopy showed that the lifetime of these species increases on going both across and down Groups 5–7.27 Indeed, CpRe(CO)2(n-heptane) was found to be the longest-lived n-heptane complex at room temperature with a lifetime of ca. 25 ms.28 The increased stability of this class of compound subsequently allowed their characterization with 1H NMR spectroscopy.29–32 Photolysis of CpRe(CO)3 in cyclopentane at 180 K yielded the corresponding alkane complex, which was the first organometallic alkane complex to be characterized by 1H NMR spectroscopy.29 A combined TRIR and NMR study has also been used to probe the interaction of alkanes with the CpRe(CO)(PF3) moiety.31 This route required the linking of a UV lamp via an optical fibre to irradiate the NMR samples in situ. The most recent example of an organometallic alkane complex to be characterized by NMR spectroscopy has come from Brookhart and co-workers who have successfully identified a rhodium-methane complex.33 Protonation of a rhodium-methyl precursor, (PONOP)Rh(CH3)+ (PONOP = 2,6-(tBu2PO)2C5H3N) in CDCl2F solvent at −110 °C afforded the corresponding rhodium-methane complex, the first such methane complex to be characterized by this technique. It is now evident that organometallic alkane complexes exhibit a proton resonance in the high-field region normally associated with hydride complexes, but these can be distinguished from one another by the large value of 1JC–H and by the isotopic perturbation of equilibrium observed on partial deuteration of the alkane.
For neutral metal carbonyl alkane complexes, the characterization of shorter-lived species in solution at low temperature by NMR spectroscopy will be greatly enhanced if one is able to generate them in high concentrations. In York, we have recently reported a combined IR and NMR study of TpRe(CO)2(c-C5H10) that built upon our earlier work using a HeCd cw laser, to integrate a pulsed Nd:YAG source into a 600 MHz wide-bore NMR spectrometer.34 Our photolysis apparatus projects light from the bottom of the spectrometer, through an air-gap in-between the probe and shim system onto the side of the NMR sample. Consequently this new approach enables a range of lasing wavelengths to be used at low temperatures with pressurised samples in sealed NMR tubes, greatly increasing the photo-conversion of starting materials to products in comparison with standard UV lamp methods. An additional advantage in achieving high concentrations of the transient can be obtained by using a precursor which has a very labile component (e.g. N2). Previous work on compounds related to the one used in this study, Cp*Re(CO)(N2)L (L = CO, phosphines or phosphites) has demonstrated that photo-induced N2 loss is a very efficient process and that such complexes are useful precursors to a range of C–H and C–Cl activation products.35–37 Photodissociation of N2 occurs with greater quantum efficiency than ejection of CO and importantly this can be achieved with longer wavelength excitation, which can also help to avoid unwanted side reactions. The use of more energetic 266 nm radiation usually required for laser NMR experiments with CpRe(CO)3 is problematic because of the high absorption coefficient (ε) at 266 nm. The high ε makes it necessary to employ solutions that are exceptionally dilute for NMR purposes; otherwise the laser radiation cannot penetrate into the centre of the tube. Precursor complexes which can be excited at lower energy where ε is lower are therefore very desirable.
In this paper, we report on an investigation of the photochemistry of CpRe(CO)2(N2) and Cp‡Re(CO)2(N2) (Cp‡ = η5-1,2-C5H3(tBu)2) in alkane solvents employing both IR and NMR spectroscopies, Scheme 1.
 | ||
| Scheme 1 Photochemical reactions of Cp/Cp‡Re(CO)2(N2) studied by a combination of time-resolved infrared and NMR spectroscopies | ||
Experimental
Materials
All synthetic manipulations were carried out under an atmosphere of N2 using standard Schlenk techniques, unless otherwise specified. All preparative reactions employed magnetic stirring, and solvents that were purified by previously described methods.38 Silica gel chromatography was performed using a Biotage SP1 purification system in open air with HPLC-grade solvents. Literature procedures were used for the preparation of 1,2-di-tert-butylcyclopentadiene,39 4-methoxybenzene diazonium tetrafluoroborate,40 and KC8.41 Re2(CO)10 was purchased from Strem and used as received. CpRe(CO)2(N2) was made according to the method of Sutton.42For the TRIR experiments, cyclopentane and n-heptane (Aldrich) were distilled over CaH2 under an argon atmosphere and degassed prior to use. CO (BOC, CP grade) was used as received. For the NMR experiments, the cyclopentane was purified before use by stirring with conc. H2SO4 overnight, washing with water and drying over MgSO4 then storing over a potassium mirror. Singly labelled carbon-13 cyclopentane, 13C-1-c-C5H10 (Aldrich) was used as received.
Time-resolved infrared spectroscopy
To obtain TRIR spectra, we have used an ultrafast TRIR apparatus, the details of which have been described elsewhere.16 Briefly, a Ti:sapphire oscillator/regenerative amplifier system (Spitfire Pro, Spectra Physics) is used to generate 800 nm laser pulses (fwhm ca. 150 fs) with an energy of 2.3 mJ at a repetition rate of 1 kHz. This is used to pump an OPA and generate broadband, tunable mid-IR pulses, with a spectral bandwidth of ca. 180 cm−1 and a pulse energy of ca. 2 μJ (at 2000 cm−1). A Q-switched Nd:YVO laser (ACE-25QSPXHP/MOPA, Advanced Optical Technology, UK) is employed as a pump source, which is synchronized to the Spitfire Pro amplifier and the delay between pump and probe pulses is controlled with a pulse generator (DG535, Stanford Research System) between 0.5 ns to 100 μs. The broadband transmitted probe pulse is detected with a 128 element MCT array detector (Infrared Associates).Room temperature TRIR kinetic measurements were obtained by monitoring the change in IR transmission at a particular frequency using a mid-IR Quantum Cascade Laser (Daylight Solutions) and a DC-coupled MCT detector (Laser Monitoring Systems Ltd), following 266 nm irradiation by a Nd:YAG laser (Spectra Physics Quanta-Ray GCR-12). A variable temperature solution cell (Harrick Corp.) with CaF2 windows (pathlength 1.5 mm) was used and the temperature of this cell was maintained at a constant 25 °C by means of a recirculating water bath. Fresh solutions of Cp‡Re(CO)2(N2) were flowed into the cell after each UV laser shot and allowed to thermally equilibrate before taking the next measurement. The solubilities of CO in n-heptane and cyclopentane‡ were taken to be 1.2 ×10−2 and 0.9 ×10−2 M at 1 atm CO and 298 K, respectively.44
The low temperature spectroscopic measurements were performed in a high pressure-low temperature (HPLT) apparatus. Briefly, a copper HPLT cell, with 5 mm thick CaF2 windows, was attached to a commercial cold-finger (CryoTiger, Brooks Automation) and a closed-cycle refrigeration system. A solution of Cp‡Re(CO)2(N2) in cyclopentane was flowed into the cell, cooled to 180 K and photolysed at 355 nm using a Nd:YAG laser (Spectra Physics Quanta-Ray, GCR-12). The subsequent reaction was monitored using a rapid-scan FTIR spectrometer (Nexus 870, Nicolet) at 4 cm−1 resolution.
Low temperature NMR spectroscopy
Results and discussion
(a) Characterization of CpRe(CO)2(alkane) in alkane solvents using a dinitrogen precursor
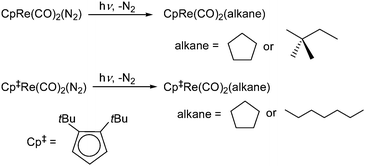
CpRe(CO)2(N2) was investigated as an alternative precursor to CpRe(CO)3 for the production of alkane complexes using the broadband light output from a 100 W mercury arc lamp coupled into a single silica fibre as described previously.29,30 It was expected that the Re–N2 bond would be more easily cleaved photolytically than the Re–CO bond and that the lifetime of the CpRe(CO)2(alkane) complexes produced may be longer as N2 is a poorer nucleophile than CO. However, formation of the Cp2Re2(CO)4(N2) dimer via reaction of the alkane complex with the starting material (or possibly Cp2Re2(CO)5via reaction with the CpRe(CO)3 impurity), is likely to be the major decomposition route. As expected, more facile N2 ligand photolysis was observed as it is susceptible to photolytic ejection at longer wavelengths than CO. This change in the precursor is significant since the requirement for components that absorb in the deep UV (e.g. pyrex glassware cannot be used), is relaxed. As a result, we found that CpRe(CO)2(N2) could be successfully photolysed in an unmodified Shigemi symmetrical NMR microtube, which contains susceptibility matched glass plugs above and below the active volume of the NMR sample. The tip of the quartz fibre was threaded down the centre of the top insert of the Shigemi tube placed against the top of the upper susceptibility matched glass plug (ca. 1 cm long). Fig. 1 shows the photolysis of CpRe(CO)2(N2) containing ca. 20% CpRe(CO)3 as an impurity in a Shigemi NMR tube in 95% c-C5H10/5% pentane-d12 at 180 K. While efficient photolysis of the CpRe(CO)2(N2) is observed, the decrease in the intensity of the CpRe(CO)3 resonance is only very slight, if present at all, throughout the course of the experiment. Formation of CpRe(CO)2(c-C5H10) is observed by production of a new resonance at δ = 4.92 in the Cp region and the characteristic quintet at δ = −2.28 in the hydride region. We find no evidence for formation of CpRe(CO)(N2)(c-C5H10). As with photolysis of CpRe(CO)3 in a normal NMR tube, clean conversion to the alkane complex is not attained, and decomposition of the product is also observed during the photolysis process. There was sufficient CpRe(CO)2(c-C5H10) produced for a 1D 13C edited experiment to be performed at natural abundance allowing the recording of a 1JC–H value of 114 (± 7) Hz observed for the bound CH2 moiety, consistent with the reported value for a 13C labelled sample (113 Hz), (see Supporting Information, Fig. S2†).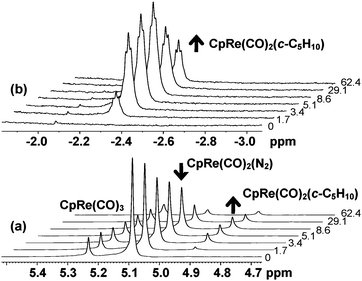 | ||
| Fig. 1 1H NMR (500 MHz) monitoring the photolysis of CpRe(CO)2(N2) containing ca 20% CpRe(CO)3 in 95% c-C5H10 /5% pentane-d12 in (a) the cyclopentadienyl and (b) the bound methylene proton region at 180 K. Each spectrum is 64 scans, processed with 1 Hz of line broadening. The formation of CpRe(CO)2(c-C5H10) is clearly observable. | ||
We have also investigated the binding of 2,2-dimethylbutane to the [CpRe(CO)2] moiety following irradiation of CpRe(CO)2(N2). Three separate shielded resonances were observed on photolysis of CpRe(CO)2(N2) in 95% 2,2-dimethylbutane/5% pentane-d12 at 170 K, corresponding to binding of the three different sites (C1 (×3), C3, C4) in the alkane to the [CpRe(CO)2] fragment, Scheme 2. Binding of the t-butyl group, CpRe(CO)2(2,2-dimethylbutane-η2-C1,H1), is dominant, giving rise to a singlet at δ = −2.19, with two other resonances at δ −1.89 (br t, 3JH-H ∼ 7.3 Hz) and δ -3.79 (br q, 3JH-H ∼ 6.5 Hz) assigned to the bound methyl in CpRe(CO)2(2,2-dimethylbutane-η2-C4,H4) and the bound methylene in CpRe(CO)2(2,2-dimethylbutane-η2-C3,H3), respectively, Fig. 2(a). Three new resonances associated with these species can be seen in the cyclopentadienyl region of the 1H NMR spectrum at δ = 4.92 – 4.95 (see Supporting Information, Fig. S3 and S4†).
![The three possible co-ordination sites of the [CpRe(CO)2] fragment along the 2,2-dimethylbutane chain.](/image/article/2010/SC/c0sc00311e/c0sc00311e-s2.gif) | ||
| Scheme 2 The three possible co-ordination sites of the [CpRe(CO)2] fragment along the 2,2-dimethylbutane chain. | ||
 | ||
| Fig. 2 (a) 1H NMR spectrum (500 MHz) resulting from photolysis of CpRe(CO)2(N2) in 95% 2,2-dimethylbutane/5% pentane-d12 at 170 K, 512 scans, processed with 3 Hz of line broadening. The new peaks are assigned to CpRe(CO)2(2,2-dimethylbutane-η2-C1,H1) (C1); CpRe(CO)2(2,2-dimethylbutane-η2-C3,H3) (C3) and CpRe(CO)2(2,2-dimethylbutane-η2-C4,H4) (C4) (b) Bottom: 1H NMR spectrum of bound methyl protons of CpRe(CO)2(2,2-dimethylbutane-η2-C1,H1); Top: same sample, 13C edited showing 1JC–H = 118 (± 5) Hz, 18,432 scans, processed with 7 Hz of line broadening. | ||
Sufficient amounts of the major isomer, CpRe(CO)2(2,2-dimethylbutane-η2-C1,H1), were present to permit a 13C edited experiment with minimal signal-to-noise ratio, which revealed a large 1JC–H value of 118 (± 5) Hz, confirming that at least the major isomer is an alkane complex, Fig. 2(b).
The use of a Hg-arc lamp as an excitation source for the NMR experiments is limited, as broadband irradiation may result in photodecomposition of the short-lived alkane complexes. The use of laser excitation at a specific wavelength can minimise these effects. In the next section we characterize a new series of alkane complexes using the related precursor, Cp‡Re(CO)2(N2) (Cp‡ = η5-1,2-C5H3(tBu)2).
(b) Characterization and reactivity of Cp‡Re(CO)2(alkane) at room temperature using TRIR spectroscopy
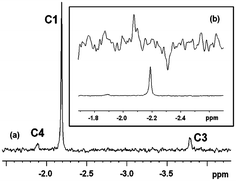
The use of the Cp‡Re(CO)2(N2) precursor for generation of organometallic alkane complexes is not straightforward since the presence of the t-butyl substituents on the Cp ring leads to the possibility that agostic complexes could be formed when a vacant site is created following photolysis. We have therefore investigated the photochemistry of Cp‡Re(CO)2(N2) in alkane solution using both fast time-resolved IR (TRIR) spectroscopy47 at room temperature and conventional IR spectroscopy at low temperature.The TRIR spectrum obtained 1 μs following photolysis (266 nm) of a solution of Cp‡Re(CO)2(N2) in cyclopentane shows bleaching of the parent ν(CO) bands at 1963 and 1908 cm−1 and the production of two new bands at lower energy than those of the parent absorptions, at 1937 and 1876 cm−1, Fig. 3(a). These new bands can be readily assigned to the formation of a species containing the [Cp‡Re(CO)2] moiety via ejection of N2, by comparison with previous TRIR studies, Table 1.24,28 However, care is needed to distinguish between [Cp‡Re(CO)2(alkane)] and a species with an agostic interaction with the t-butyl groups. Oxidative addition to form a species [Cp‡Re(CO)2(H)(R)] where R = alkyl or cyclometallation) can be excluded by the shift to low wavenumber of the product ν(CO) bands, with respect to the precursor. This result can be directly compared to the analogous CpRe(CO)2(CH4) and CpRe(CO)2(CH3)H complexes where the shift to higher wavenumber of the ν(CO) bands is indicative of the formation of the alkyl hydride complex.15 Close inspection of the spectra shows no evidence for the loss of CO, indicating that N2 loss is the major photochemical pathway. The formation of this species following irradiation of Cp‡Re(CO)2(N2) can be directly compared to the formation of CpRe(CO)2(alkane) from photoinduced CO loss from CpRe(CO)3, under similar conditions.24 The photoejection of N2 is more efficient with a ca. 4-fold increase in the yield of the dicarbonyl versus the CO loss pathway (see Supporting Information, Fig. S5†). The loss of N2 can also be accomplished using lower energy UV excitation. Fig. 3(b) shows the TRIR difference spectrum 1 μs after 355 nm photolysis of the same solution. It exhibits the same features as those obtained after 266 nm photolysis although the yield of the dicarbonyl is considerably lower due to the lower UV extinction coefficient at this excitation wavelength. Nevertheless, the use of 355 nm light does lead to the efficient formation of a Re-dicarbonyl species (through loss of N2), which is not possible with CpRe(CO)3.
 | ||
| Fig. 3 TRIR difference spectra of Cp‡Re(CO)2(N2) in cyclopentane at 298 K 1 μs after photolysis at (a) 266 nm, (b) 355 nm and (c) FTIR difference spectrum 50 ms after 355 nm photolysis at 180 K. | ||
| Complex | ν(CO)/cm−1 | Conditions | Ref. |
|---|---|---|---|
| a ν(NN) at 2138 and 2131 in both n-heptane and cyclopentane at 298 and 180 K. | |||
| (η5-C5H5)Re(CO)3 | 2029, 1934 | Nujol, 77 K | 48 |
| 2031, 1940 | n-Heptane, 298 K | 28 | |
| 2030, 1938 | n-Heptane, 190 K | 24 | |
| 2031, 1939.5 | Cyclopentane, 298 K | 24 | |
| 2029, 1936.5 | Cyclopentane, 180 K | 24 | |
| (η5-C5Me5)Re(CO)3 | 2013, 1930 | Cyclopentane, 180 K | 24 |
| (η5-C5Ph5)Re(CO)3 | 2013, 1930 | Cyclopentane, 180 K | 24 |
| (η5-1,2-C5H3(tBu)2)Re(CO)2(N2) a | 1963, 1909 | n-Heptane, 298 K | This Work |
| 1962, 1908 | Cyclopentane, 298 K | This Work | |
| 1961, 1905 | Cyclopentane, 180 K | This Work | |
| (η5-C5H5)Re(CO)2 | 2029, 1934 | Nujol, 77 K | 48 |
| (η5-C5H5)Re(CO)2(n-C7H16) | 1945, 1890 | n-Heptane, 298 K | 28 |
| (η5-C5H5)Re(CO)2(n-C7H16) | 1948, 1883 | n-Heptane, 190 K | 24 |
| (η5-C5H5)Re(CO)2(c-C5H10) | 1951, 1886 | Cyclopentane, 298 K | 24 |
| (η5-C5H5)Re(CO)2(c-C5H10) | 1947.5, 1881.5 | Cyclopentane, 180 K | 24 |
| (η5-C5Me5)Re(CO)2(c-C5H10) | 1928.5, 1864 | Cyclopentane, 180 K | 24 |
| (η5-C5Ph5)Re(CO)2(c-C5H10) | 1943, 1881 | Cyclopentane, 180 K | 24 |
| (η5-1,2-C5H3(tBu)2)Re(CO)2(n-C7H16) | 1940, 1876 | n-Heptane, 298 K | This Work |
| (η5-1,2-C5H3(tBu)2)Re(CO)2(c-C5H10) | 1940, 1877 | Cyclopentane, 298 K | This Work |
| (η5-1,2-C5H3(tBu)2)Re(CO)2(c-C5H10) | 1939, 1873 | Cyclopentane, 180 K | This Work |
In order to investigate whether [Cp‡Re(CO)2] is stabilised by an agostic interaction between the t-butyl groups on the Cp ring and the Re centre, or exists as an alkane complex, additional experimental evidence was recorded. In the presence of CO (2 atm) [Cp‡Re(CO)2] decays to form CpRe(CO)3 with a lifetime of 8.4 ms (kobs = 118 (± 5) s−1, at 25 °C), Fig. 4(a). The second-order rate constant for the reaction between [Cp‡Re(CO)2] and CO, kCO, was measured in two different alkane solvents, cyclopentane and n-heptane. Previous experiments have demonstrated that cyclic alkanes tend to give longer-lived alkane complexes compared to their linear counterparts.25,26 This phenomenon has been associated with differences in the activation entropy (ΔS‡) of the reactions of these two types of complexes. Indeed, experiments on the related species, CpRe(CO)2(alkane) have shown that the cyclopentane complex is less reactive towards CO than the n-heptane complex by a factor of approximately 2.24 For the decay of the N2 loss intermediate, [Cp‡Re(CO)2], a plot of the pseudo first-order rate constant, kobs, as a function of the concentration of CO at 25 °C, yields the second-order rate constant, kCO, Fig. 5.
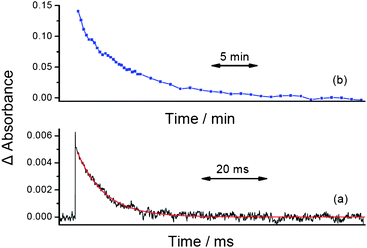 | ||
| Fig. 4 Kinetic traces showing the decay of Cp‡Re(CO)2(c-C5H10) in cyclopentane at (a) 25 °C, in the presence of CO (2 atm) and (b) at 180 K. | ||
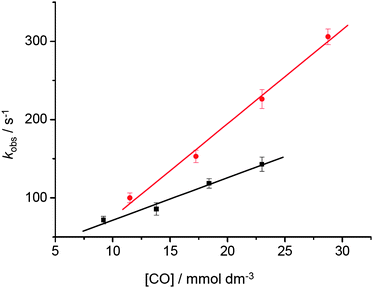 | ||
| Fig. 5 Plot of kobsversus CO concentration for the decay of Cp‡Re(CO)2(alkane) at 25 °C in (•) n-heptane and (■) cyclopentane. The gradient of these plots yields the second-order rate constant, kCO. | ||
The values of kCO for the reaction of [Cp‡Re(CO)2] in n-heptane and cyclopentane are 1.2 (± 0.1) ×104 and 5.4 (± 0.3) ×103 dm3 mol−1 s−1, respectively. These data suggest that there is no significant formation of an agostic interaction between the t-butyl groups on the Cp ring and the Re centre, and allow us to assign the N2 loss intermediate as Cp‡Re(CO)2(alkane), which is confirmed by the NMR spectroscopic work detailed below. A comparison of the rates of reaction between Cp and Cp‡ rhenium alkane complexes is presented in Table 2. Substitution of hydrogen on the cyclopentadienyl ring by t-butyl groups increases the reactivity of the corresponding alkane complex (n-heptane or cyclopentane) around 5 times. This can be explained by steric interference between the alkane and the cyclopentadienyl ring. Previous work on related half-sandwich organometallic alkane compounds, such as (η5-C5R5)M(CO)2(alkane) (R = H, Me, Et or Ph; M = Mn or Re), 23,24 and Cr(η6-C6R6)(CO)2(alkane) (R = H, Me or Et)26 has shown that steric rather than electronic effects significantly influence the reactivity of organometallic alkane complexes.
(c) Characterization and reactivity of Cp‡Re(CO)2(c-C5H10) at 180 K using TRIR spectroscopy
To investigate the behaviour of Cp‡Re(CO)2(N2) in alkane solution at low temperature, a sample was prepared in cyclopentane and cooled to 180 K. Photolysis at 355 nm leads to bleaching of the parent IR bands (1961 and 1905 cm−1) and the formation of two new bands at lower energy (1939 and 1873 cm−1), Fig. 3(c). At 180 K, the bands due to the cyclopentane complex, Cp‡Re(CO)2(c-C5H10) decay with a lifetime of 5.5 min (kobs = 3.0 (± 0.3) ×10−3 s−1), Fig. 4(b). This system is more reactive than the previous Re-alkane complexes that have been detected by NMR spectroscopy but we considered that it was still sufficiently long-lived to be detected by the latter technique.(d) Characterization of Cp‡Re(CO)2(c-C5H10) at 190 K by NMR spectroscopy
The 1H NMR spectrum of Cp‡Re(CO)2(N2) in neat cyclopentane at 190 K recorded with solvent suppression shows two proton signals of the cyclopentadienyl ligand at δ = 5.09 (d, 3JH–H = 3.0 Hz) and 4.67 (t, 3JH–H = 2.9 Hz), which integrate to 2H and 1H, respectively. The resonance of the t-butyl group is obscured by the solvent resonance. Laser irradiation (355 nm) of this solution results in a decrease in intensity of these resonances and the appearance of two new Cp resonances at δ = 5.00 (d, 3JH–H = 3.0 Hz) and 4.53 (t, 3JH–H = 2.8 Hz), together with a new broad multiplet in the high-field region at δ = −2.44, Fig. 6. These three new resonances can be assigned to Cp‡Re(CO)2(c-C5H10) by comparison with previous studies of CpRe(CO)2(c-C5H10) and TpRe(CO)2(c-C5H10).29,34 The resonance at δ = −2.44 arises from one methylene unit of the cyclopentane ring which is interacting with the metal centre. The measured lifetime (t½) of the alkane complex obtained from the NMR results is 10.6 min (kobs 1.57 (± 0.15) × 10−3 s−1) at 190 K based on data collected after 10 min of photolysis, (see Supporting Information, Fig. S6†). This value is slightly larger than that obtained from low temperature FTIR measurements.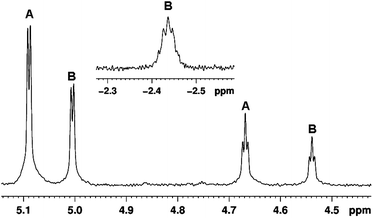 | ||
| Fig. 6 Low-field region of the 1H NMR spectrum obtained after 355 nm laser irradiation of Cp‡Re(CO)2(N2) (resonances labelled A) in cyclopentane at 190 K in the Cp region of the spectrum showing formation of Cp‡Re(CO)2(c-C5H10) (B); (inset) resonance due to the co-ordinated hydrogen of the cyclopentane in Cp‡Re(CO)2(c-C5H10). A Gaussian function was used to improve spectral resolution. | ||
In order to demonstrate that the peak at δ = −2.44 originates from the co-ordination of the solvent, rather than from an agostic interaction of the t-butyl groups on the Cp ligand or from a hydride ligand, additional experiments were performed using mono-deuterated cyclopentane, c-C5H9D. Photolysis of Cp‡Re(CO)2(N2) in neat c-C5H9D resulted in the production of two new resonances in the bound-alkane region of the 1H NMR spectrum at δ = −2.43 (m) and −4.20, with an intensity pattern in the ratio of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, Fig. 7.
:1, Fig. 7.
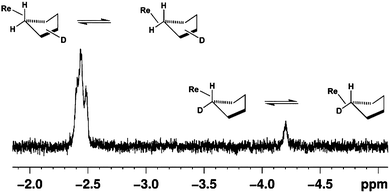 | ||
| Fig. 7 High-field region of the 1H NMR spectrum obtained after 355 nm laser irradiation of Cp‡Re(CO)2(N2) in c-C5H9D at 190 K. | ||
This isotopic perturbation of equilibrium (IPE) can be explained if one co-ordinated C–H bond is undergoing rapid exchange with the unbound geminal proton of the interacting methylene unit of the alkane, but precludes a rapid walk around the carbon framework. This observation parallels those made in previous studies of Re-alkane complexes,29,30,34 and confirms that the solvent is the source of the resonance at δ = − 2.43. The more intense resonance (δ = −2.43) occurs in the same position as that observed for c-C5H10 and corresponds to isotopomers with one deuterium on the unco-ordinated carbon atoms (Cβ and Cγ).34,49 The resonance at δ = −4.20 corresponds to isotopomers with one deuterium on the co-ordinated carbon (Cα). The value of the shift Δδ is 1.77 ppm, compared to 1.80 ppm for the Cp analogue.27
A similar experiment was also carried out using neat mono-carbon-13 labelled cyclopentane, 13C-1-c-C5H10 as the solvent. After photolysis, the 1H NMR spectrum displays the expected resonance at δ = −2.44 flanked by satellites with a large 1JC–H coupling of 114 Hz (i.e. the same as for the Cp analogue). This value is smaller than that found in free cyclopentane (129.4 Hz) by about 12%.29 The 13C chemical shift was determined by 1H-13C correlation as δ = −33.8 (Fig. 8). These values demonstrate unambiguously the formation of a weakly bound σ-alkane complex by comparison with previous examples.25–27,29
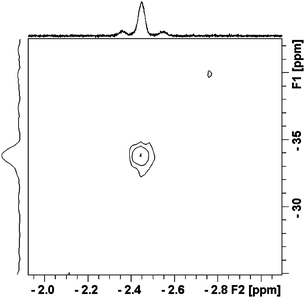 | ||
| Fig. 8 1H-13C HMQC NMR spectrum of Cp‡Re(CO)2(c-C5H10) after photolysis at 355 nm in neat 13C-1-c-C5H10 at 190 K. Note the satellites on the proton resonance. | ||
Conclusions
In this paper, we have demonstrated the advantages of employing dinitrogen complexes, CpRe(CO)2(N2) and Cp‡Re(CO)2(N2), as precursors for the formation of organometallic alkane complexes. The dinitrogen complex allows for the use of lower energy irradiation and gives a higher product yield. The lower absorption coefficient of the precursor complexes at 355 nm permits better matching of concentrations for photolysis (within the NMR tube) and NMR detection. Despite the electron donation from the t-butyl groups, the alkane complex is more reactive towards CO than its unsubstituted analogue, probably because of steric hindrance. The characteristic features of large isotopic shift of resonance on deuteration of the alkane, large 1JC–H, and a large negative 13C NMR chemical shift allow us to demonstrate conclusively that Cp‡Re(CO)2(c-C5H10) is an alkane complex and that no agostic species are present. As for CpRe(CO)2(c-C5H10), the deuteration experiments on Cp‡Re(CO)2(c-C5H10) show that there is rapid exchange between the η2-CH and its geminal neighbour, but that there is no evidence for exchange with the other hydrogens of cyclopentane, indicating that chain walking is slow on the NMR timescale. We have also shown that CpRe(CO)2(2,2-dimethylbutane) may be formed from CpRe(CO)2(N2) as three isomers differing in their co-ordination to either of the CH3 or CH2 moieties.We have employed two alternative technical approaches to in situ photolysis: use of a microtube in conjunction with a UV lamp and optical fibre and use of a conventional NMR tube in conjunction with laser irradiation. For IR spectroscopy, we have made use of fast TRIR methods at room temperature and FTIR spectroscopy at low temperature, such that the conditions of measuring the IR and NMR spectra are very similar.
Acknowledgements
We thank the EPSRC (EP/D058031 and EP/D055768) and the Universities of Nottingham and York for funding. We wish to thank Mr. P. Fields, Mr. R. Wilson, Mr. M. Guyler and Dr D. C. Williamson for technical assistance. MWG gratefully acknowledges receipt of a Royal Society Wolfson Merit Award. OT would like to thank the Ministerio de Ciencia e Innovación and the Fundación Española para la Ciencia y la Tecnología for funding. PV and RGB acknowledge initial support by the Director, Office of Energy Research, Office of Basic Energy Sciences, Chemical Sciences Division, of the U.S. Department of Energy, under Contract DE-AC02-05CH11231, and then (for PV) by the NSF (CHE-0907800). GB thanks the Australian Research Council for financial support.Notes and references
- G. J. Kubas, Metal Dihydrogen and σ-Bond Complexes, Kluwer Academic Publishers, New York, 2001 Search PubMed.
- R. N. Perutz and C. Hall, Chem. Rev., 1996, 96, 3125–3146 CrossRef CAS.
- J. J. Schneider, Angew. Chem., Int. Ed. Engl., 1996, 35, 1068–1075 CrossRef CAS.
- R. G. Bergman, Nature, 2007, 446, 391–393 CrossRef CAS.
- A. J. Cowan and M. W. George, Coord. Chem. Rev., 2008, 252, 2504–2511 CrossRef CAS.
- R. H. Crabtree, J. Organomet. Chem., 2004, 689, 4083–4091 CrossRef CAS.
- M. Lersch and M. Tilset, Chem. Rev., 2005, 105, 2471–2526 CrossRef CAS.
- W. D. Jones, Inorg. Chem., 2005, 44, 4475–4484 CrossRef.
- R. N. Perutz and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2007, 46, 2578–2592 CrossRef CAS.
- R. N. Perutz and J. J. Turner, J. Am. Chem. Soc., 1975, 97, 4791–4800 CrossRef CAS.
- M. Poliakoff and J. J. Turner, J. Chem. Soc., Dalton Trans., 1974, 2276–2285 RSC.
- J. M. Kelly, H. Hermann and E. K. von Gustorf, J. Chem. Soc., Chem. Commun., 1973, 105–106 RSC.
- H. Hermann, F. W. Grevels, A. Henne and K. Schaffner, J. Phys. Chem., 1982, 86, 5151–5154 CrossRef CAS.
- M. W. George, M. T. Haward, P. A. Hamley, C. Hughes, F. P. A. Johnson, V. K. Popov and M. Poliakoff, J. Am. Chem. Soc., 1993, 115, 2286–2299 CrossRef CAS.
- A. J. Cowan, P. Portius, H. K. Kawanami, O. S. Jina, D. C. Grills, X. Z. Sun, J. McMaster and M. W. George, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6933–6938 CrossRef CAS.
- M. A. H. Alamiry, N. M. Boyle, C. M. Brookes, M. W. George, C. Long, P. Portius, M. T. Pryce, K. L. Ronayne, X.-Z. Sun, M. Towrie and K. Q. Vuong, Organometallics, 2009, 28, 1461–1468 CrossRef CAS.
- M. Besora, J.-L. Carreon-Macedo, A. J. Cowan, M. W. George, J. N. Harvey, P. Portius, K. L. Ronayne, X.-Z. Sun and M. Towrie, J. Am. Chem. Soc., 2009, 131, 3583–3592 CrossRef CAS.
- J. A. Calladine, K. Q. Vuong, X. Z. Sun and M. W. George, Pure Appl. Chem., 2009, 81, 1667–1675 CrossRef CAS.
- S. T. Belt, F. W. Grevels, W. E. Klotzbücher, A. McCamley and R. N. Perutz, J. Am. Chem. Soc., 1989, 111, 8373–8382 CrossRef CAS.
- R. H. Schultz, A. A. Bengali, M. J. Tauber, B. H. Weiller, E. P. Wasserman, K. R. Kyle, C. B. Moore and R. G. Bergman, J. Am. Chem. Soc., 1994, 116, 7369–7377 CrossRef CAS.
- S. E. Bromberg, H. Yang, M. C. Asplund, T. Lian, B. K. McNamara, K. T. Kotz, J. S. Yeston, M. J. Wilkens, H. Frei, R. G. Bergman and C. B. Harris, Science, 1997, 278, 260–263 CrossRef CAS.
- A. J. Blake, M. W. George, M. B. Hall, J. McMaster, P. Portius, X. Z. Sun, M. Towrie, C. E. Webster, C. Wilson and S. D. Zaric, Organometallics, 2008, 27, 189–201 CrossRef CAS.
- F. P. A. Johnson, M. W. George, V. N. Bagratashvili, L. N. Vereshchagina and M. Poliakoff, Mendeleev Commun., 1991, 1, 26–28 Search PubMed.
- G. I. Childs, C. S. Colley, J. Dyer, D. C. Grills, X. Z. Sun, J. X. Yang and M. W. George, J. Chem. Soc., Dalton Trans., 2000, 1901–1906 RSC.
- C. J. Breheny, J. M. Kelly, C. Long, S. O'Keeffe, M. T. Pryce, G. Russell and M. M. Walsh, Organometallics, 1998, 17, 3690–3695 CrossRef CAS.
- B. S. Creaven, M. W. George, A. G. Ginzburg, C. Hughes, J. M. Kelly, C. Long, I. M. McGrath and M. T. Pryce, Organometallics, 1993, 12, 3127–3131 CrossRef CAS.
- G. I. Childs, D. C. Grills, X. Z. Sun and M. W. George, Pure Appl. Chem., 2001, 73, 443–447 CrossRef CAS.
- X.-Z. Sun, D. C. Grills, S. M. Nikiforov, M. Poliakoff and M. W. George, J. Am. Chem. Soc., 1997, 119, 7521–7525 CrossRef CAS.
- S. Geftakis and G. E. Ball, J. Am. Chem. Soc., 1998, 120, 9953–9954 CrossRef CAS.
- D. J. Lawes, S. Geftakis and G. E. Ball, J. Am. Chem. Soc., 2005, 127, 4134–4135 CrossRef CAS.
- G. E. Ball, C. M. Brookes, A. J. Cowan, T. A. Darwish, M. W. George, H. K. Kawanami, P. Portius and J. P. Rourke, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6927–6932 CrossRef CAS.
- D. J. Lawes, T. A. Darwish, T. Clark, J. B. Harper and G. E. Ball, Angew. Chem., Int. Ed., 2006, 45, 4486–4490 CrossRef CAS.
- W. H. Bernskoetter, C. K. Schauer, K. I. Goldberg and M. Brookhart, Science, 2009, 326, 553–556 CrossRef CAS.
- S. B. Duckett, M. W. George, O. S. Jina, S. L. Matthews, R. N. Perutz, X. Z. Sun and K. Q. Vuong, Chem. Commun., 2009, 1401–1403 RSC.
- A. H. Klahn-Oliva, R. D. Singer and D. Sutton, J. Am. Chem. Soc., 1986, 108, 3107–3108 CrossRef.
- R. Arancibia, F. Godoy, M. T. Garland, A. Ibanez, R. Baggio and A. H. Klahn, J. Organomet. Chem., 2007, 692, 963–967 CrossRef CAS.
- J. J. Carbó, O. Eisenstein, C. L. Higgitt, A. H. Klahn, F. Maseras, B. Oelckers and R. N. Perutz, J. Chem. Soc., Dalton Trans., 2001, 1452–1461 RSC.
- P. J. Alaimo, D. W. Peters, J. Arnold and R. G. Bergman, J. Chem. Educ., 2001, 78, 64 CrossRef CAS.
- R. P. Hughes, A. S. Kowalski, J. R. Lomprey and D. R. Neithamer, J. Org. Chem., 1996, 61, 401–404 CrossRef CAS.
- M. R. Heinrich, O. Blank, D. Ullrich and M. Kirschstein, J. Org. Chem., 2007, 72, 9609–9616 CrossRef CAS.
- J.-M. Lalancette, G. Rollin and P. Dumas, Can. J. Chem., 1972, 50, 3058–3062 CAS.
- A. Cusanelli and D. Sutton, Organometallics, 1995, 14, 4651–4660 CrossRef CAS.
- R. P. Hughes and J. R. Lomprey, Inorg. Chim. Acta, 1995, 240, 653–656 CrossRef CAS.
- R. W. Cargill, ed., IUPAC Solubility Data Series Vol. 43 Carbon Monoxide, Pergamon Press, Oxford, 1990 Search PubMed.
- M. L. Lui, X. A. Mao, C. H. Ye, H. Huang, J. K. Nicholson and J. C. Lindon, J. Magn. Reson., 1998, 132, 125–129 CrossRef CAS.
- C. Godard, P. Callaghan, J. L. Cunningham, S. B. Duckett, J. A. B. Lohman and R. N. Perutz, Chem. Commun., 2002, 2836–2837 RSC.
- M. W. George, M. Poliakoff and J. J. Turner, Analyst, 1994, 119, 551 RSC.
- T. E. Bitterwolf, K. A. Lott, A. J. Rest and J. Mascetti, J. Organomet. Chem., 1991, 419, 113–126 CrossRef CAS.
- R. B. Calvert and J. R. Shapley, J. Am. Chem. Soc., 1978, 100, 7726–7727 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Diagram of set-up for laser photolysis in NMR probe, 13C edited 1H NMR spectrum of CpRe(CO)2(cyclopentane), 1H NMR spectrum of CpRe(CO)2(2,2-dimethylbutane), TRIR traces showing production of CO and N2 loss intermediates and a plot showing the lifetime of Cp‡Re(CO)2(cyclopentane) measured by 1H NMR spectroscopy. See DOI: 10.1039/c0sc00311e |
| ‡ To our knowledge the solubility of CO in cyclopentane has not been measured, and so we have assumed it to be the same as for cyclohexane. |
| This journal is © The Royal Society of Chemistry 2010 |