N-Heterocyclic carbene-catalyzed tandem aza-benzoin/Michael reactions: on site reversal of the reactivity of N-Boc imines†‡
Ke-Jia
Wu
,
Gong-Qiang
Li
,
Yi
Li
,
Li-Xin
Dai
and
Shu-Li
You
*
State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Lu, Shanghai 200032, P. R. China. E-mail: slyou@mail.sioc.ac.cn; Fax: (+86) 21-54925087
First published on 18th October 2010
Abstract
A tandem NHC-catalyzed aza-benzoin/Michael reaction has been developed as a method to efficiently produce dihydroindenones and pyrrolidinone-containing tricycles. The novel reaction pattern involves tert-butyl aryl(tosyl)methylcarbamates reacting as both electrophile and nucleophile on the same carbon.
Imines and their precursors serve as equivalents of carbonyl compounds and represent one of the most important classes of intermediates used in organic synthesis.1 Owing to their ready preparation and high stability, tert-butyl aryl(tosyl)methylcarbamates have been developed as precursors of N-Boc imines, thus greatly expanding the utility of imines in synthesis.2 In general, imines and their precursors participate as electrophilic acceptors in reactions with various nucleophiles. In recent studies of N-heterocyclic carbene (NHC)-catalyzed umpolung reactions,3,4 we observed that imines behave as suitable acyl anion acceptors.5 In efforts aimed at the development of a new strategy for the facile construction of the dihydroisoquinolinone scaffold, we designed and explored a tandem NHC-catalyzed aza-benzoin/aza-Michael reaction (Scheme 1).
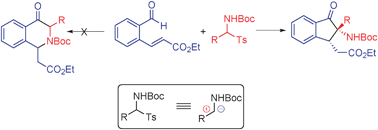 | ||
| Scheme 1 Tandem aza-benzoin/Michael reaction. | ||
Surprisingly, the process was observed to follow an unconventional tandem aza-benzoin/Michael pathway that produces substituted dihydroindenones in a highly diastereoselective manner. In this process, tert-butyl aryl(tosyl)methylcarbamate acts as both an electrophile and nucleophile at the same carbon site. Importantly, the substituted dihydroindenones generated in this process can be readily converted to pharmaceutically interesting pyrrolidinone-containing tricyclic substances that contain two stereogenic centers including one quaternary carbon.6 To our knowledge, no reports exist describing this type of reactivity profile for imines. Below, we describe the results of this exploratory study.
The current study began with an exploration of the reaction of (E)-ethyl 3-(2-formylphenyl)acrylate 1a with tert-butyl (4-methoxyphenyl)(tosyl)methylcarbamate 2a, which provided the results summarized in Table 1. In the presence of thiazolium salt 4 (10 mol%) and Et3N (2.2 equiv.), reaction of 1a and 2a did not produce the aza-benzoin/Michael reaction product 3aa but instead afforded the aza-benzoin product 3aa′ in 62% yield (Table 1, entry 1). We envisaged that use of a stronger base would facilitate the subsequent Michael addition reaction of 3aa′. Screening various bases (Table 1, entries 1–4) led to the identification of DBU as a suitable base for the tandem reaction, which generated 3aa in 31% yield with an excellent dr (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The yield of this process was improved to 68% when Cs2CO3 was employed as the base (Table 1, entry 6). It is worth noting that the use of a reduced loading of Cs2CO3 (1.5 equiv.) led to formation of a mixture of 3aa and 3aa′ in a ratio of 1:3, while increasing the Cs2CO3 loading (4 equiv.) resulted in a lower yield (Table 1, entries 7, 8). A probe of reactions promoted by several catalysts under these conditions demonstrated that thiazolium salt 4 was optimal (see the SI for details). An examination of various solvents (toluene, CH3CN, ether, CH2Cl2, dioxane, t-BuOH, and THF) revealed that CH2Cl2 was optimal (Table 1, entries 6, 9–15). Specifically, reaction of 1a with 2a in CH2Cl2 gave 3aa in 76% yield with a high diastereoselectivity (dr >20:1). The relative stereochemistry of 3aa was determined after the subsequent cyclization (vide infra).
:1). The yield of this process was improved to 68% when Cs2CO3 was employed as the base (Table 1, entry 6). It is worth noting that the use of a reduced loading of Cs2CO3 (1.5 equiv.) led to formation of a mixture of 3aa and 3aa′ in a ratio of 1:3, while increasing the Cs2CO3 loading (4 equiv.) resulted in a lower yield (Table 1, entries 7, 8). A probe of reactions promoted by several catalysts under these conditions demonstrated that thiazolium salt 4 was optimal (see the SI for details). An examination of various solvents (toluene, CH3CN, ether, CH2Cl2, dioxane, t-BuOH, and THF) revealed that CH2Cl2 was optimal (Table 1, entries 6, 9–15). Specifically, reaction of 1a with 2a in CH2Cl2 gave 3aa in 76% yield with a high diastereoselectivity (dr >20:1). The relative stereochemistry of 3aa was determined after the subsequent cyclization (vide infra).
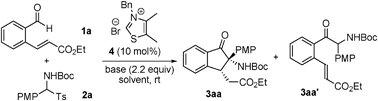
| Entry | Base | Solvent | Time/h | 3aa, yield (%)b | 3aa, dr | 3aa′, yield (%)b |
|---|---|---|---|---|---|---|
| a Reaction conditions: 1a/2a/base = 2.0/1.0/2.2, 0.1 M of 2a in solvent at rt. b Isolated yields. c 1.5 equiv. of Cs2CO3 was used. d 4.0 equiv. of Cs2CO3 was used. | ||||||
| 1 | Et3N | CHCl3 | 8 | — | — | 62 |
| 2 | DIPEA | CHCl3 | 8 | — | — | 63 |
| 3 | DABCO | CHCl3 | 8 | — | — | 34 |
| 4 | DBU | CHCl3 | 5 | 31 | >20:1 |
— |
| 5 | K2CO3 | CHCl3 | 8 | — | — | 71 |
| 6 | Cs2CO3 | CHCl3 | 5 | 68 | >20:1 |
— |
| 7c | Cs2CO3 | CHCl3 | 5 | 18 | >20:1 |
51 |
| 8d | Cs2CO3 | CHCl3 | 5 | 59 | >20:1 |
— |
| 9 | Cs2CO3 | Toluene | 5 | Trace | — | — |
| 10 | Cs2CO3 | CH3CN | 5 | 46 | >20:1 |
— |
| 11 | Cs2CO3 | Et2O | 5 | 19 | >20:1 |
— |
| 12 | Cs2CO3 | CH2Cl2 | 5 | 76 | >20:1 |
— |
| 13 | Cs2CO3 | Dioxane | 5 | 41 | >20:1 |
— |
| 14 | Cs2CO3 | t-BuOH | 5 | N.R. | — | — |
| 15 | Cs2CO3 | THF | 5 | 47 | >20:1 |
— |
Reactions under the optimized reaction conditions (10 mol% thiazolium salt 4, 220 mol% Cs2CO3, in CH2Cl2, rt) of a variety of substituted (E)-ethyl 3-(2-formylphenyl)acrylates and N-Boc imine precusors were explored in order to examine the generality of the tandem reaction. As the results displayed in Table 2 show, the tert-butyl aryl(tosyl)methylcarbamates 2a–f, which contain either electron-donating (4-MeO, 2-MeO, 4-Me, and 3-Me) or electron-withdrawing (4-Br and 4-Cl) groups, participate in the tandem reaction to afford the desired products 3 in 76–93% yields (Table 2, entries 1–6, 9). In addition, heteroaryl (2-furyl and 2-thienyl) and 1-naphthyl-derived imine precursors also serve as suitable substrates in this process (62–90% yields, Table 2, entries 7–8, 10). Unfortunately, reaction of the alkyl-substituted substrate 2k failed to produce an adduct under these conditions (Table 2, entry 11). In the (E)-ethyl 3-(2-formylphenyl)acrylate series, methyl (1b) and fluoro groups (1c) are tolerated (Table 2, entries 12–13). Finally, it should be noted that the preformed N-Boc imine could also be used in this reaction although the yield is slightly decreased (Table 2, entry 14).
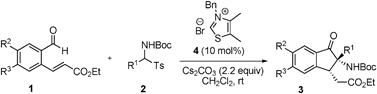
| Entry | 1, R2, R3 | 2, R1 | 3, yield (%)b | dr |
|---|---|---|---|---|
| a Reaction conditions: 1/2/base = 2.0/1.0/2.2, 0.1 M of 2 in CH2Cl2 at rt. b Isolated yields. c The preformed imine was used. | ||||
| 1 | 1a, H, H | 2a, 4-MeOC6H4 | 3aa, 76 | >20:1 |
| 2 | 1a, H, H | 2b, 2-MeOC6H4 | 3ab, 83 | >20:1 |
| 3 | 1a, H, H | 2c, 4-MeC6H4 | 3ac, 85 | >20:1 |
| 4 | 1a, H, H | 2d, 3-MeC6H4 | 3ad, 93 | >20:1 |
| 5 | 1a, H, H | 2e, 4-BrC6H4 | 3ae, 82 | >20:1 |
| 6 | 1a, H, H | 2f, 4-ClC6H4 | 3af, 81 | >20:1 |
| 7 | 1a, H, H | 2g, 2-furyl | 3ag, 62 | >20:1 |
| 8 | 1a, H, H | 2h, 2-thienyl | 3ah, 79 | >20:1 |
| 9 | 1a, H, H | 2i, phenyl | 3ai, 77 | >20:1 |
| 10 | 1a, H, H | 2j, 1-naphthyl | 3aj, 90 | >20:1 |
| 11 | 1a, H, H | 2k, c-C6H13 | N.R. | — |
| 12 | 1b, Me, H | 2i, phenyl | 3bi, 52 | >20:1 |
| 13 | 1c, H, F | 2i, phenyl | 3ci, 89 | >20:1 |
| 14c | 1a, H, H |
|
3ae, 62 | >20:1 |
A plausible catalytic cycle for this process is depicted in Scheme 2. N-Heterocyclic carbene I, generated by base induced deprotonation of the corresponding thiazolium salt, reacts with aldehyde 1a to produce the Breslow intermediate II.7 Intermediate IV is then formed by reaction of II with the electrophilic intermediate III, generated from 2i under the basic conditions. Proton transfer from intermediate IV gives intermediate V, which releases the carbene catalyst I and the key α-amino ketone intermediate 3ai′. The propensity to form an enolate makes the α-carbon in 3ai′ a stronger nucleophilic site than the amine nitrogen and drives the exclusive formation of the intramolecular Michael addition product 3ai rather than the intramolecular aza-Michael addition product.
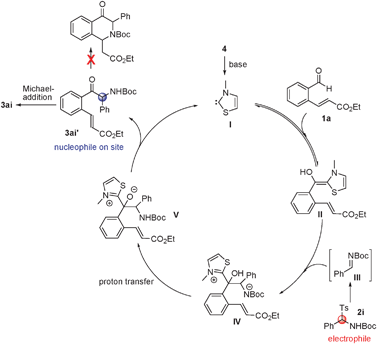 | ||
| Scheme 2 Proposed mechanism for the tandem reaction. | ||
The multi-functionalized products obtained in the aza-benzoin/Michael reactions have the potential of participating in a variety of transformations. For instance, upon treatment with TFA at room temperature, the adducts 3 are converted to the pyrrolidinone-containing tricyclic products 5 in excellent yields (77% to 91%) via a sequence involving deprotection of the Boc group followed by intramolecular lactamization (Table 3).
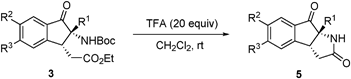
| Entry | 3, R1, R2, R3 | 5, yield (%)b |
|---|---|---|
| a Reaction conditions: 3/TFA = 1.0/20.0, 0.03 M of 3 in CH2Cl2 at rt. b Isolated yields. | ||
| 1 | 3aa, 4-MeOC6H4, H, H | 5aa, 82 |
| 2 | 3ab, 2-MeOC6H4, H, H | 5ab, 77 |
| 3 | 3ac, 4-MeC6H4, H, H | 5ac, 86 |
| 4 | 3ad, 3-MeC6H4, H, H | 5ad, 79 |
| 5 | 3ae, 4-BrC6H4, H, H | 5ae, 85 |
| 6 | 3af, 4-ClC6H4, H, H | 5af, 90 |
| 7 | 3ag, 2-furyl, H, H | 5ag, 91 |
| 8 | 3ah, 2-thienyl, H, H | 5ah, 86 |
| 9 | 3ai, phenyl, H, H | 5ai, 80 |
| 10 | 3aj, 1-naphthyl, H, H | 5aj, 81 |
| 11 | 3bi, phenyl, Me, H | 5bi, 87 |
| 12 | 3ci, phenyl, H, F | 5ci, 81 |
The structure of the pyrrolidinone-containing product 5ae was confirmed by using X-ray crystallographic analysis (Fig. 1).8 The tricyclic substances 5 are not only pharmaceutically interesting but they also represent a synthetically challenging family of compounds owing to the presence of two stereogenic centers including one quaternary carbon.
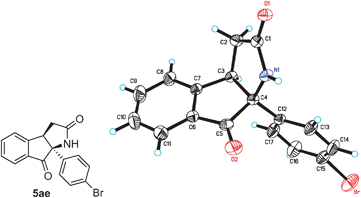 | ||
| Fig. 1 X-ray crystal structure of pyrrolidinone-containing tricycle 5ae (thermal ellipsoids are set at 30% probability). | ||
In summary, the studies described above have led to the development of an efficient, highly diastereoselective, tandem NHC-catalyzed aza-benzoin/Michael reaction affording dihydroindenones that can be transformed to pyrrolidinone-containing tricycles. This is the first report of a process in which the N-Boc imine precursor, tert-butyl aryl(tosyl)methylcarbamate, acts as both an electrophile and nucleophile at the same carbon site. We believe that the interesting structural features of the products of this new reaction along with the novelty of the reaction pattern of imines will make the results of this effort particularly interesting to synthetic organic chemists.
We thank the National Natural Science Foundation of China (20732006, 20821002, 20972177) and National Basic Research Program of China (973 Program 2009CB825300) for generous financial support.
Notes and references
- For reviews: (a) M. Petrini and E. Torregiani, Synthesis, 2007, 159 CrossRef CAS; (b) G. K. Friestad and A. K. Mathies, Tetrahedron, 2007, 63, 2541 CrossRef CAS; (c) D. Ferraris, Tetrahedron, 2007, 63, 9581 CrossRef CAS.
- Selected recent examples: (a) E. Gomez-Bengoa, A. Linden, R. López, I. Múgica-Mendiola, M. Oiarbide and C. Palomo, J. Am. Chem. Soc., 2008, 130, 7955 CrossRef CAS; (b) C. Gianelli, L. Sambri, A. Carlone, G. Bartoli and P. Melchiorre, Angew. Chem., Int. Ed., 2008, 47, 8700 CrossRef CAS; (c) M. Terada, K. Machioka and K. Sorimachi, Angew. Chem., Int. Ed., 2009, 48, 2553 CrossRef CAS.
- For reviews: (a) D. Enders and T. Balensiefer, Acc. Chem. Res., 2004, 37, 534 CrossRef CAS; (b) J. S. Johnson, Angew. Chem., Int. Ed., 2004, 43, 1326 CrossRef CAS; (c) M. Christmann, Angew. Chem., Int. Ed., 2005, 44, 2632 CrossRef CAS; (d) K. Zeitler, Angew. Chem., Int. Ed., 2005, 44, 7506 CrossRef CAS; (e) N. Marion, S. Díez-González and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2988 CrossRef CAS; (f) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606 CrossRef CAS; (g) V. Nair, S. Vellalath and B. P. Babu, Chem. Soc. Rev., 2008, 37, 2691 RSC; (h) W. D. Jones, J. Am. Chem. Soc., 2009, 131, 15075 CrossRef CAS.
- (a) G.-Q. Li, Y. Li, L.-X. Dai and S.-L. You, Org. Lett., 2007, 9, 3519 CrossRef CAS; (b) Y. Li, Z. Feng and S.-L. You, Chem. Commun., 2008, 2263 RSC; (c) G.-Q. Li, Y. Li, L.-X. Dai and S.-L. You, Adv. Synth. Catal., 2008, 350, 1258 CrossRef CAS; (d) Y. Li, Z.-A. Zhao, H. He and S.-L. You, Adv. Synth. Catal., 2008, 350, 1885 CrossRef CAS; (e) G.-Q. Li, L.-X. Dai and S.-L. You, Org. Lett., 2009, 11, 1623 CrossRef CAS; (f) Y. Li, F.-Q. Shi, Q.-L. He and S.-L. You, Org. Lett., 2009, 11, 3182 CrossRef CAS; (g) Y. Li, X.-Q. Wang, C. Zheng and S.-L. You, Chem. Commun., 2009, 5823 RSC.
- (a) G.-Q. Li, L.-X. Dai and S.-L. You, Chem. Commun., 2007, 852 RSC. Also see reports from other groups: (b) J. Castells, F. López-Calahorra, M. Bassedas and P. Urrios, Synthesis, 1988, 314 CrossRef CAS; (c) J. A. Murry, D. E. Frantz, A. Soheili, R. Tiller, E. J. J. Grabowski and P. J. Reider, J. Am. Chem. Soc., 2001, 123, 9696 CrossRef CAS; (d) D. E. Frantz, L. Morency, A. Soheili, J. A. Murry, E. J. J. Grabowski and R. D. Tillyer, Org. Lett., 2004, 6, 843 CrossRef CAS; (e) W. Li and Y. Lam, J. Comb. Chem., 2005, 7, 644 CrossRef CAS; (f) S. M. Mennen, J. D. Gipson, Y. R. Kim and S. J. Miller, J. Am. Chem. Soc., 2005, 127, 1654 CrossRef CAS.
- (a) P. A. Crooks, F. DeSimone and E. Ramundo, J. Heterocycl. Chem., 1982, 19, 1433 CrossRef CAS; (b) P. A. Crooks, T. Deeks and F. DeSimone, J. Heterocycl. Chem., 1989, 26, 1113 CrossRef CAS; (c) Y. L. Chen, J. Nielsen, K. Hedberg, A. Dunaiskis, S. Jones, L. Russo, J. Johnson, J. Ives and D. Liston, J. Med. Chem., 1992, 35, 1429 CrossRef CAS; (d) E. Gavuzzo and M. Pomponi, J. Biochem. Mol. Toxicol., 2002, 16, 64 CrossRef CAS; (e) K. S. Feldman and M. R. Iyer, J. Am. Chem. Soc., 2005, 127, 4590 CrossRef CAS.
- R. Breslow, J. Am. Chem. Soc., 1958, 80, 3719 CrossRef CAS.
- CCDC 772193 contains the supplementary crystallographic data for 5ae.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures and analysis data for new compounds, CIF file of 5ae. CCDC 772193. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0cc01769h |
| This journal is © The Royal Society of Chemistry 2011 |