Synthesis and self-assembly of poly(3-hexylthiophene)-block-poly(acrylic acid)†‡
Zicheng
Li
,
Robert J.
Ono
,
Zong-Quan
Wu
and
Christopher W.
Bielawski
*
Department of Chemistry and Biochemistry, The University of Texas at Austin, 1 University Station A5300, Austin, TX 78712, USA. E-mail: bielawski@cm.utexas.edu
First published on 28th September 2010
Abstract
A modular and convenient synthesis of ethynyl end functionalized poly(3-hexylthiophene) in high purity is reported; this material facilitated access to poly(3-hexylthiophene)-block-poly(acrylic acid) which self-assembled into hierarchical structures.
Donor–acceptor block copolymers containing regioregular poly(3-hexylthiophene) (P3HT) have garnered considerable attention in recent years as promising materials for applications in optoelectronics.1 Such copolymers are often prepared using a grafting-from approach, where an end-functionalized polythiophene is used as a macroinitiator for the chain extension polymerization of a second block.2 A significant drawback to this strategy, however, is the need for multiple post-polymerization modifications as the requisite initiator must be installed onto the end of the P3HT chain. A convenient alternative is to use a grafting-to approach, whereby the constituent homopolymers are independently synthesized and then subsequently linked together.
Because the utility of the grafting-to method rests on the efficiency by which the chain-ends can react with each other, a reaction is needed that is both high-yielding and functional group tolerant. A transformation that fulfills these criteria is the Cu-catalyzed “click” azide–alkyne cycloaddition,3 which has found remarkable utility in the field of polymer science,4 including the synthesis of P3HT-containing block copolymers. For example, Urien et al. synthesized a series of P3HT-containing di- and triblock copolymers from alkyne end-functionalized P3HT and azide end-functionalized polystyrenes.5 More recently, Segalman et al. reported a block copolymer where an ethynyl-terminated P3HT was “clicked” with a polyacrylate that was grown from an azido-functionalized initiator.6
While these seminal reports highlight the potential of using grafting methods to synthesize P3HT-based block copolymers, the materials obtained often contain inseparable homopolymer impurities that impede copolymer formation.§ A simple method that effectively overcomes these issues and rapidly affords appreciable quantities of P3HT-containing block copolymers in high yield and free of homopolymer impurities remains an important challenge in synthetic polymer chemistry, especially within the context of optoelectronic applications, where materials of high purity are needed.
Herein we report a convenient synthesis of P3HT-containing rod–coil diblock copolymersvia the “click” reaction of ethynyl-terminated P3HT7 (P3HT–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH) and azide-terminated poly(tert-butyl acrylate)8 (PtBA–N3). Central to our results is the discovery that P3HT–CCH is unstable and engages in homocoupling over time. This limitation was effectively surmounted with the development of an improved isolation procedure which facilitated the synthesis of pure samples of P3HT–CCH and strongly influenced the outcome of its subsequent cycloaddition reactivity. The choice of PtBA as the coil block enabled access to the amphiphilic diblock copolymer, P3HT-block-poly(acrylic acid) (P3HT-b-PAA), which was recently shown9 by McCullough et al. to exhibit solvatochromic behavior in a variety of polar and non-polar solvents.¶ Prompted by this report and as part of a newly launched program aimed at studying donor–acceptor copolymers that adopt highly ordered structures, we also disclose the characterization of the self-assembly behavior of P3HT-b-PAA using dynamic light scattering (DLS) and transmission electron microscopy (TEM).
CH) and azide-terminated poly(tert-butyl acrylate)8 (PtBA–N3). Central to our results is the discovery that P3HT–CCH is unstable and engages in homocoupling over time. This limitation was effectively surmounted with the development of an improved isolation procedure which facilitated the synthesis of pure samples of P3HT–CCH and strongly influenced the outcome of its subsequent cycloaddition reactivity. The choice of PtBA as the coil block enabled access to the amphiphilic diblock copolymer, P3HT-block-poly(acrylic acid) (P3HT-b-PAA), which was recently shown9 by McCullough et al. to exhibit solvatochromic behavior in a variety of polar and non-polar solvents.¶ Prompted by this report and as part of a newly launched program aimed at studying donor–acceptor copolymers that adopt highly ordered structures, we also disclose the characterization of the self-assembly behavior of P3HT-b-PAA using dynamic light scattering (DLS) and transmission electron microscopy (TEM).
As summarized in Scheme 1, PtBA–Br with different chain lengths were synthesized viaCu-mediated atom transfer radical polymerization (65 °C, neat) by varying the initial tert-butyl acrylate to initiator (ethyl 2-bromoisobutyrate; EBiB) ratios. After precipitation from a methanol/water mixture (1/1, v/v), the desired polymers were isolated viafiltration and then characterized by gel permeation chromatography (GPC) as well as 1H NMR spectroscopy (see Table 1; entries 1–3). Displacement of the bromide end-group was accomplished with NaN3 in DMF (50 °C), which afforded the respective azide-functionalized PtBA (PtBA–N3) in 85–90% isolated yields after washing the products with water.8 Incorporation of the azide group was confirmed by 1H NMR spectroscopy through the observation of an upfield shift in the signal attributed to the terminal methine from δ = 4.05 (CH–Br) to 3.78 ppm (CH–N3) (CDCl3), and by IR spectroscopy from the characteristic frequency observed at νN3 = 2119 cm−1 (KBr).
 | ||
| Scheme 1 Synthesis of P3HT-b-PtBA and P3HT-b-PAA. | ||
| Entry | Polymer a | [M]0/[I]0b | Isolated yield (%) | M n c/Da | M w/Mnc |
|---|---|---|---|---|---|
| a The subscripted numbers denote the respective homopolymer's degree of polymerization, as determined by GPC. b Initial monomer (tBA or 2,5-dibromo-3-hexylthiophene) to initiator (EBiB or Ni(dppp)Cl2) (dppp = 1,3-bis(diphenylphosphino)propane) ratio. c M n and Mw/Mn were determined by GPC and are reported as their polystyrene equivalents. | |||||
| 1 | PtBA57N3 | 80/1 | 87 | 7400 | 1.27 |
| 2 | PtBA112N3 | 120/1 | 85 | 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 650 650 |
1.32 |
| 3 | PtBA170N3 | 300/1 | 85 | 22000 |
1.32 |
| 4 |
P3HT29CCH
|
50/1 | 60 | 4900 | 1.24 |
| 5 |
P3HT96CCH
|
100/1 | 50 | 16200 |
1.14 |
| 6 |
P3HT138CCH
|
150/1 | 52 | 23300 |
1.23 |
| 7 | P3HT96-b-PtBA57 | — | 77 | 24100 |
1.22 |
| 8 | P3HT96-b-PtBA112 | — | 72 | 32000 |
1.24 |
| 9 | P3HT96-b-PtBA170 | — | 66 | 41400 |
1.25 |
| 10 | P3HT29-b-PtBA112 | — | 60 | 24900 |
1.40 |
| 11 | P3HT138-b-PtBA112 | — | 69 | 42000 |
1.32 |
Regioregular ethynyl-terminated P3HT (P3HT–CCH) was prepared from 2,5-dibromo-3-hexylthiophene and isopropylmagnesium chloride10via a Ni-catalyzed Grignard metathesis (GRIM) polymerization according to literature procedures.5,7 After allowing the reaction to proceed for 10 min at room temperature, ethynylmagnesium bromide was added which simultaneously installed an ethynyl end-group and quenched the polymerization. Using this method, a variety of P3HT–CCH homopolymers were synthesized by adjusting the initial monomer to catalyst ratio. Following precipitation from methanol, the materials were isolated in 50–60% yields by filtration (Table 1; entries 4–6). Analysis of the isolated polymers by GPC showed narrow molecular weight distributions characteristic of GRIM polymerizations, and the incorporation of the ethynyl end-group was confirmed by 1H NMR spectroscopy where a signal at δ = 3.52 ppm (CDCl3) was observed, consistent with literature values.11
During the course of the aforementioned syntheses, it was discovered that P3HT–CCH was highly sensitive to the isolation and purification conditions employed. For example, subjecting the polymer to sequential Soxhlet extractions—a standard protocol7 for the purification of P3HTs, including P3HT–CCH—we observed the gradual growth of a high molecular weight material that corresponded to nearly twice that of the bulk of the P3HT product, as determined by GPC (Fig. 1A). Neat samples of P3HT–CCH left on the benchtop under ambient conditions for extended periods of time (>1 d) exhibited similar behavior.‡ The origin of these high molecular weight polymers was attributed to alkyne–alkyne homocoupling reactions catalyzed by residual metal catalyst.|| Accordingly, an improved purification procedure that eliminated the use of Soxhlet extractions altogether was developed: upon conclusion of the aforementioned GRIM polymerization reactions and addition of ethynylmagnesium bromide, the corresponding mixtures were poured into excess methanol and the precipitated polymers were collected viafiltration, and then washed with excess methanol and hexanes. As shown in Fig. 1B, this method afforded pure, monomodal samples of P3HT–CCH that were free of high molecular weight impurities.**
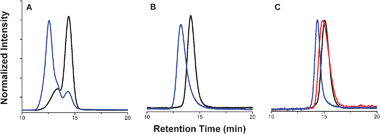 | ||
| Fig. 1 Representative GPC traces of (A) P3HT–CCH purified using standard methodology (black) and its corresponding P3HT-b-PtBA copolymer (blue) obtained using UV-vis detection at 450 nm; (B) P3HT96–CCH purified using the methodology reported herein (black) and its corresponding P3HT96-b-PtBA170 copolymer (blue) obtained using UV-vis detection at 450 nm; and (C) P3HT96–CCH purified using the methodology reported herein (black), PtBA170–N3homopolymer (red), and their corresponding P3HT96-b-PtBA170 copolymer (blue) obtained using refractive index detection. GPC conditions: 25 °C, THF as eluent. | ||
With the aforementioned homopolymers in hand, efforts shifted toward linking these materials. Under the optimized reaction conditions, equimolar amounts of P3HT–CCH and PtBA–N3 were combined with two equiv. of N,N,N′,N″,N″-pentamethyldiethylenetriamine (PMDETA)/CuBr as the catalyst system,4,5 and then stirred for 24 h at 40 °C in THF. After filtering the resulting reaction mixture through neutral alumina (eluent = THF) to remove the catalyst, the desired P3HT-b-PtBA block copolymers were obtained in 60–77% isolated yields by precipitating the reaction mixtures into methanol. Signals attributable to both coupling partners along with the disappearance of the signal assigned to the alkynyl moiety (δ = 3.52 ppm, CDCl3) were observed by 1H NMR spectroscopy and were accompanied by disappearance of the diagnostic νN3IR signal. Moreover, the GPC traces of the isolated copolymers were monomodal with narrow distributions (Fig. 1C) and correlated well with their expected molecular weights (see Table 1, entries 7–11).
Building on the synthesis of pure P3HT-b-PtBA copolymers, we shifted our attention toward exploring amphiphilic derivatives. Acidolysis of the P3HT-b-PtBA block copolymers was performed according to literature procedures,9 using an excess of trifluoroacetic acid (TFA) in CHCl3 and afforded P3HT-b-PAA in high yield (97%). The disappearance of the 1H NMR signal attributed to the tert-butyl group in P3HT-b-PtBA (δ = 1.40 ppm; THF-d8) accompanied by the presence of the –OH stretch at ∼3400 cm−1 in the IR spectrum was consistent with a complete deprotection reaction.
Slowly adding an equal volume of water (a good solvent for PAA and a poor solvent for P3HT) to a stirred THF solution of P3HT-b-PAA (initial conc. = 1.0 mg mL−1) at room temperature afforded a purple solution that was accompanied by a bathochromic shift (433 → 507 nm) in the UV/vis absorption maxima.‡ This behavior was consistent with the well-established solvatochromism of regioregular P3HT,9,12 and was in agreement with the color change observed13 during the formation of P3HT-encapsulated micelles. Following dialysis against de-ionized water to remove the residual THF, visually transparent aqueous solutions were obtained. As shown in Fig. 2, TEM revealed that spherical micellar nanostructures with narrow size distributions were formed for P3HT96-b-PAA170 and P3HT29-b-PAA112 (Table 1; entries 9 and 10, respectively). The micelles assembled from the former exhibited larger average diameters (Dav) (28.5 ± 4.4 nm) than those prepared from the latter (Dav = 17.2 ± 5.3 nm), consistent with the difference in molecular weights of the constituent homopolymers. Intensity-averaged hydrodynamic diameters (Dh) of the nanoparticles, as determined by DLS, exhibited a similar trend, with P3HT96-b-PAA170 having a Dh of 63.6 ± 2.2 nm and P3HT29-b-PAA112 a Dh of 37.5 ± 2.4 nm.
 | ||
| Fig. 2 TEM images of micelles (stained with 1 wt% aqueous solution of phosphotungstic acid) formed from P3HT96-b-PAA170 (left) and P3HT29-b-PAA112 (right) (scale bar = 100 nm). | ||
In conclusion, we have prepared P3HT-b-PtBA copolymersvia coupling of ethynyl-terminated P3HT with azide-terminated PtBA. Access to pristine P3HT–CCH was found to be critical for the success of the aforementioned reaction and a convenient method for cleanly isolating this polymer was developed. The amphiphilic block copolymer P3HT-b-PAA was obtained upon acidolysis of P3HT-b-PtBA and found to self-assemble into micellar structures with sizes dependent on copolymer molecular weight and composition. We believe that the method reported herein will facilitate the creation and development of new block copolymers of high purity that are suitable for use and study in a broad range optoelectronic and semiconducting devices.
The material presented is based upon work supported as part of the program “Understanding Charge Separation and Transfer at Interfaces in Energy Materials (EFRC:CST),” an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0001091.
Notes and references
- K. Sivula, Z. T. Ball, N. Watanabe and J. M. J. Fréchet, Adv. Mater., 2006, 18, 206 CrossRef CAS; C. Yang, J. K. Lee, A. J. Heeger and F. Wudl, J. Mater. Chem., 2009, 19, 5416 RSC; F. Richard, C. Brochon, N. Leclerc, D. Eckhardt, T. Heiser and G. Hadziioannou, Macromol. Rapid Commun., 2008, 29, 885 CrossRef CAS; Q. Zhang, A. Cirpan, T. P. Russell and T. Emrick, Macromolecules, 2009, 42, 1079 CrossRef CAS; J. U. Lee, A. Cirpan, T. Emrick, T. P. Russell and W. H. Jo, J. Mater. Chem., 2009, 19, 1483 RSC; J.-H. Tsai, Y.-C. Lai, T. Higashihara, C.-J. Lin, M. Ueda and W.-C. Chen, Macromolecules, 2010, 43, 6085 CrossRef CAS.
- J. Liu, E. Sheina, T. Kowalewski and R. D. McCullough, Angew. Chem., Int. Ed., 2002, 41, 329 CrossRef CAS; M. C. Iovu, M. Jeffries-EL, E. E. Sheina, J. R. Cooper and R. D. McCullough, Polymer, 2005, 46, 8582 CrossRef CAS; M. C. Iovu, C. R. Craley, M. Jeffries-EL, A. B. Krankowski, R. Zhang, T. Kowalewski and R. D. McCullough, Macromolecules, 2007, 40, 4733 CrossRef CAS; C.-A. Dai, W.-C. Yen, Y.-H. Lee, C.-C. Ho and W.-F. Su, J. Am. Chem. Soc., 2007, 129, 11036 CrossRef CAS; G. A. Mussie, G. Srinivas, S. John and C. S. Mihaela, Macromol. Chem. Phys., 2009, 210, 2007 CrossRef CAS; B. W. Boudouris, C. D. Frisbie and M. A. Hillmyer, Macromolecules, 2010, 43, 3566 CrossRef CAS; I. Botiz and S. B. Darling, Macromolecules, 2009, 42, 8211 CrossRef CAS; C. P. Radano, O. A. Scherman, N. Stingelin-Stutzmann, C. Muller, D. W. Breiby, P. Smith, R. A. J. Janssen and E. W. Meijer, J. Am. Chem. Soc., 2005, 127, 12502 CrossRef CAS.
- C. K. Hartmuth, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- J.-F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018 CrossRef CAS; W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15 CrossRef CAS; M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963 CrossRef CAS; B. S. Sumerlin and A. P. Vogt, Macromolecules, 2010, 43, 1 CrossRef CAS; P. L. Golas and K. Matyjaszewski, QSAR Comb. Sci., 2007, 26, 1116 Search PubMed; A. E. Richard, Aust. J. Chem., 2007, 60, 384 CrossRef CAS.
- M. Urien, H. Erothu, E. Cloutet, R. C. Hiorns, L. Vignau and H. Cramail, Macromolecules, 2008, 41, 7033 CrossRef CAS.
- Y. Tao, B. McCulloch, S. Kim and R. A. Segalman, Soft Matter, 2009, 5, 4219 RSC.
- M. Jeffries-EL, G. Sauve and R. D. McCullough, Macromolecules, 2005, 38, 10346 CrossRef CAS.
- L. Qingchun and C. Yongming, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6103 CrossRef CAS.
- C. R. Craley, R. Zhang, T. Kowalewski, R. D. McCullough and M. C. Stefan, Macromol. Rapid Commun., 2009, 30, 11 CrossRef CAS.
- R. Miyakoshi, A. Yokoyama and T. Yokozawa, J. Am. Chem. Soc., 2005, 127, 17542 CrossRef CAS; R. S. Loewe, P. C. Ewbank, J. Liu, L. Zhai and R. D. McCullough, Macromolecules, 2001, 34, 4324 CrossRef CAS.
- T. L. Benanti, A. Kalaydjian and D. Venkataraman, Macromolecules, 2008, 41, 8312 CrossRef CAS.
- U. Scherf, A. Gutacker and N. Koenen, Acc. Chem. Res., 2008, 41, 1086 CrossRef CAS.
- M. Wang, S. Kumar, A. Lee, N. Felorzabihi, L. Shen, F. Zhao, P. Froimowicz, G. D. Scholes and M. A. Winnik, J. Am. Chem. Soc., 2008, 130, 9481 CrossRef CAS.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Additional experimental procedures, spectroscopic data, gel permeation chromatograms, and DLS details. See DOI: 10.1039/c0cc02166k |
| § For example, Urien et al. reported5 that P3HT–CCH was deactivated and did not react with azide-terminated polystyrenes. In light of the results presented herein which also employ P3HT–CCH under nearly identical reaction conditions, we believe that the quality of this polymer is critical to determining the outcome of its cycloaddition with azides. |
| ¶ The P3HT-b-PAA studied by McCullough et al. was prepared using a grafting-from approach where poly(tBA) was grown from a functionalized P3HT macroinitiator.9 |
| || In support of this hypothesis, trimethylsilyl protected ethynyl terminated P3HT (P3HT–CC–TMS) was found to be stable to ambient conditions for indefinite periods of time. |
| ** Storage of purified P3HT–CCH was found to form high molecular weight impurities over time. It is recommended that the material be used immediately upon preparation, or stored under an inert atmosphere in a freezer (−20 °C). |
| This journal is © The Royal Society of Chemistry 2011 |