Stabilizing self-assembled Fmoc–F5–Phe hydrogels by co-assembly with PEG-functionalized monomers†‡
Derek M.
Ryan
,
Todd M.
Doran
and
Bradley L.
Nilsson
*
Department of Chemistry, University of Rochester, Rochester, NY, 14627-0216, USA. E-mail: nilsson@chem.rochester.edu; Fax: +1 585 276-0205; Tel: +1 585 276-3053
First published on 8th October 2010
Abstract
Hydrogels derived from self-assembled Fmoc–F5–Phe fibrils are stabilized with respect to shear-response by co-assembly with C-terminal PEG-functionalized Fmoc–F5–Phe.
Hydrogels formed from the dynamic self-assembly of peptides and amino acids have found use in regenerative medicine, drug delivery, and material fabrication.1,2 While peptides have been very effective hydrogelators, small molecule systems are attractive due to cost and synthetic customizability.1Fluorenylmethoxycarbonyl (Fmoc) protected amino acids and short peptides in particular self-assemble to form hydrogels similar in rheological strength to those formed from much longer peptides and covalently cross-linked polymers.3–6
We recently reported that halogenation of the phenyl side chain of Fmoc–Phe resulted in rapid self-assembly and hydrogelation relative to Fmoc–Phe or Fmoc–Tyr.7,8 Hydrogels formed from the self-assembly of Fmoc–F5–Phe–OH or monohalogenated derivatives were mechanically rigid, but solvolytically unstable. It was found that the fibrillar network precipitated after extended resting times (1–2 weeks) or immediately after the application of stress, resulting in the loss of gel properties; reformation of the gel network could not be induced once fibrils were desolvated. This phenomenon was hypothesized to be due to the low water solubility of Fmoc–F5–Phe–OH-derived fibrils. In order for hydrogels to be conveniently used in medicinal and biological applications, they must exhibit shear responsive behavior, spontaneously recovering, or healing, following the application of stress.9 Thus, even though Fmoc–F5–Phe–OH rapidly self-assembles and forms rigid hydrogels, it has limited potential in biological applications since the gel network would not survive the process of injection that has been shown to be ideal for delivery of bioactive hydrogels in vivo.9
Existing self-assembled hydrogel systems that exhibit shear-responsive behavior are derived from fibrils that are highly water soluble.9 In these systems, the application of stress temporarily disrupts the entangled fibril network, yet the fibrils themselves remain solvated, facilitating reformation of the hydrogel network when stress is removed.9 Based on these principles we hypothesized that slightly increasing the water solubility of Fmoc–F5–Phe–OH would solubilize the fibrils and impart stabilizing and shear responsive behavior to these small molecule gels. The addition of polyethyleneglycol (PEG) chains has been shown to increase the solution phase stability of fibrils in the context of amyloid forming peptides.10–12 We therefore synthesized Fmoc–F5–Phe–PEG (Fig. 1, see ESI‡ for details) in order to investigate the influence of PEG functionality on the self-assembly and stability of the resulting fibrillar network.
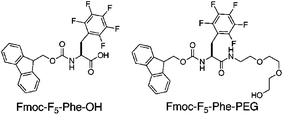 | ||
| Fig. 1 Structures of Fmoc–F5–Phe–OH and Fmoc–F5–Phe–PEG. | ||
Hydrogels were prepared by dilution of DMSO solutions of monomer into water. Fmoc–F5–Phe–PEG (Fig. 1) was dissolved in DMSO (247 mM) and was then diluted in water to a final concentration of 4.9 mM (2% DMSO/H2O).8 Upon dilution into water, an opaque suspension was formed which, when left undisturbed, became optically clear after 3–4 hours. We previously found that this transition coincides with the formation of ordered fibrillar assemblies. This transition time is much longer for Fmoc–F5–Phe–PEG than for Fmoc–F5–Phe–OH, which requires only minutes to undergo assembly and hydrogel formation. Heating the opaque suspension of Fmoc–F5–Phe–PEG to 100 °C accelerated this transition from hours to minutes. Upon cooling, a self-supporting hydrogel formed as indicated by vial inversion. In comparison, heating Fmoc–F5–Phe–OH suspensions resulted in precipitation of the material from solution. These results suggest that while incorporation of PEG does in fact increase the water solubility of the assembled network, it significantly slows the rate of assembly, possibly due to the steric influence of the C-terminal PEG functionality. To confirm that the observed optical transition was in fact due to the self-assembly of Fmoc–F5–Phe–PEG into fibrils, a circular dichroism (CD) spectrum was obtained (Fig. 2A). Fibers derived from Fmoc protected aromatic amino acids have been shown to have characteristic CD signatures at 270–310 nm, and 200–210 nm corresponding to the n–π* Fmoc–Fmoc excitation and π–π* phenyl side chain excitations, respectively.7 The magnitude of the CD signal of Fmoc–F5–Phe–PEG was significantly weaker than the corresponding signal of assembled Fmoc–F5–Phe–OH at the same concentration (Fig. 2A). In addition, the CD signals were inverted relative to Fmoc–F5–Phe–OH, appearing as maxima rather than minima. This result could indicate that Fmoc–F5–Phe–PEG is merely dissolving in water and not self-assembling into fibrils.
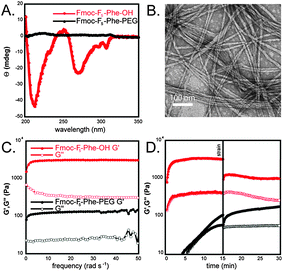 | ||
| Fig. 2 (A) CD spectra of assembled Fmoc–F5–Phe–OH (red) and Fmoc–F5–Phe–PEG (black). (B) TEM image of fibrils formed from the assembly of Fmoc–F5–Phe–PEG. (C) Dynamic frequency sweep of the gels formed from Fmoc–F5–Phe–OH (red) and Fmoc–F5–Phe–PEG (black). (D) Dynamic time sweep of Fmoc–F5–Phe–OH (red) and Fmoc–F5–Phe–PEG before and after the application of 100% strain for 30 seconds. | ||
In order to probe this possibility, transmission electron microscopy (TEM) images of Fmoc–F5–Phe–PEG gels were obtained. These TEM images clearly indicate the presence of abundant ordered fibrillar assemblies with diameters of 15 ± 1 nm (Fig. 2B). In some cases helical tapes were observed (Fig. S1, ESI‡), perhaps arising from inhibition of bundling by the C-terminal PEG groups. Based on our working model for the packing of Fmoc–F5–Phe–OH (see Fig. S2, ESI‡) we hypothesized that the rather sterically demanding PEG chain perturbs this mode of assembly, explaining the observed differences in the respective CD spectra.
Once it was established that Fmoc–F5–Phe–PEG was assembled into fibrillar networks, the rheological strength of the resulting hydrogels was measured by an oscillatory frequency sweep from 0–50 rad s−1 with 0.2% strain (Fig. 2C). Rheology was collected in the linear viscoelastic region (Fig. S3, ESI‡). Fmoc–F5–Phe–PEG hydrogels have a G′ and G″ of 130 ± 60 Pa and 30 ± 10 Pa, compared to Fmoc–F5–Phe–OH (G′ = 3060 ± 70 Pa, G″ = 320 ± 10 Pa). In addition, the G′ and G″ of Fmoc–F5–Phe–PEG displayed a large frequency dependence at oscillatory frequencies greater than 50 rad s−1, consistent with the formation of a viscous solution, while the G′ and G″ of the Fmoc–F5–Phe–OH hydrogel was essentially independent of frequency consistent with a rigid hydrogel.13 (Fig. 2C). These results demonstrate that the addition of PEG to Fmoc–F5–Phe–OH, while an efficient method for promoting water solubility of the assembled fibril network, has a detrimental effect on the rheological strength of the resulting fibrillar network.
In order to test the effect of PEG on the mechanical stability of the hydrogels, a dynamic time sweep (conducted at 0.2% strain) was performed on freshly prepared gels of Fmoc–F5–Phe–OH and Fmoc–F5–Phe–PEG. Once the G′ had plateaued, indicating completion of assembly, the samples were subjected to 100% strain for 30 seconds in order to disrupt the gel networks. Immediately following the application of 100% strain, a second time sweep was performed (0.2% strain) in order to monitor the ability of the samples to recover from the stress (Fig. 2D). It was observed that Fmoc–F5–Phe–OH only recovered 66% of its maximum G′ and was no longer a rigid hydrogel.13 More importantly, post strain samples of Fmoc–F5–Phe–OH gels showed the presence of abundant fibrillar precipitates (Fig. S4, ESI‡). In contrast, the Fmoc–F5–Phe–PEG gels fully recovered after cessation of 100% strain, demonstrating that while Fmoc–F5–Phe–PEG fibrils form weak gels, they do display shear recovery. In fact gels of Fmoc–F5–Phe–PEG were found to be fully solvated after more than 4 weeks, while Fmoc–F5–Phe–OH hydrogels were only stable for 1–2 weeks, after which precipitation was observed.
Based on these observations we hypothesized that co-assembly of Fmoc–F5–Phe–PEG and Fmoc–F5–Phe–OH would result in rigid hydrogels that exhibit shear recovery ability ideal for delivery by injection. It is known that the addition of complementary impurities to a co-assembled gel network serves to slow precipitation by introducing structural heterogeneity that slows crystal growth.14 It was hypothesized that the statistical distribution of the Fmoc–F5–Phe–PEG throughout the fibrils, would solubilize the fibrillar network relative to Fmoc–F5–Phe–OH alone.
The co-assembly behavior of Fmoc–F5–Phe and Fmoc–F5–Phe–PEG was studied in order to probe this hypothesis. Stock solutions (DMSO) were prepared with varying ratios of Fmoc–F5–Phe–OH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶Fmoc–F5–Phe–PEG (1∶1, 4∶1, and 9∶1; 247 mM total monomer concentration). Each mixture was then diluted to a final concentration of 4.9 mM total monomer in water (2% DMSO/H2O). The 1∶1 mixture formed an opaque suspension immediately after the addition of water, and required more than 3 h to become optically clear, similar to the transition time observed for the pure Fmoc–F5–Phe–PEG suspension. The 4∶1 and 9∶1 mixtures both formed opaque solutions following the addition of water; however, each became optically clear after ∼30 minutes. The resulting hydrogels were found to be stable to vial inversion, visually indicating the formation of rigid hydrogels.
∶Fmoc–F5–Phe–PEG (1∶1, 4∶1, and 9∶1; 247 mM total monomer concentration). Each mixture was then diluted to a final concentration of 4.9 mM total monomer in water (2% DMSO/H2O). The 1∶1 mixture formed an opaque suspension immediately after the addition of water, and required more than 3 h to become optically clear, similar to the transition time observed for the pure Fmoc–F5–Phe–PEG suspension. The 4∶1 and 9∶1 mixtures both formed opaque solutions following the addition of water; however, each became optically clear after ∼30 minutes. The resulting hydrogels were found to be stable to vial inversion, visually indicating the formation of rigid hydrogels.
The gels were analyzed by CD spectroscopy in order to investigate if each possessed the characteristic features associated with fibrillar assemblies of Fmoc–F5–Phe–OH (Fig. 3A). It was observed that each of the mixtures had CD signatures commonly associated with Fmoc–F5–Phe–OH fibrils; however, there was an apparent change in sign of the signal with the varying ratios.7 The 4∶1 mixture possessed a signal with the same sign as the Fmoc–F5–Phe–OH hydrogels (minima at 215 nm, 270–310 nm), while the 1∶1 and 9∶1 mixtures have the same spectral signatures as maxima. This change in sign implies there may be a potential change in helicity of the fibrils relative to Fmoc–F5–Phe–OH.8 Each mixture was then imaged by TEM in order to see if there were any observable morphological differences in the samples that could account for the observed change in CD signal (Fig. 3B–D). All three mixtures were comprised of densely entangled fibrils with average diameters of 16 ± 2 nm, 14 ± 1 nm, and 16 ± 2 nm for the 1∶1, 4∶1, and 9∶1 mixtures. While the average diameters of the bundles were similar in dimension, the handedness of individual helical fibrils was not clearly distinguishable and further structural characterization is needed in order to address the observed changes in CD spectra. These spectra are interpreted as evidence of Fmoc–Fmoc, phenyl–phenyl transitions, supporting the structural model shown in Fig. S2, ESI‡.7,8
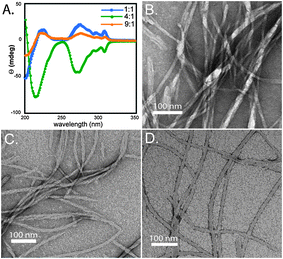 | ||
| Fig. 3 (A) CD spectra of Fmoc–F5–Phe–OH∶Fmoc–F5–Phe–PEG co-fibrils (1∶1, blue; 4∶1, green; 9∶1, orange). (B) TEM image of fibrils observed in 1∶1 Fmoc–F5–Phe–OH∶Fmoc–F5–Phe–PEG mixture. (C) TEM image of fibrils observed in 4∶1 Fmoc–F5–Phe–OH∶Fmoc–F5–Phe–PEG mixture. (D) TEM image of fibrils observed in 9∶1 Fmoc–F5–Phe–OH∶Fmoc–F5–Phe–PEG mixture. | ||
The rheological strength of each of the hydrogels was measured using a dynamic frequency sweep from 0–50 rad s−1 with 0.2% strain (Fig. 4A). The G′ and G″ of the 1∶1 co-fibrils displayed significant frequency dependence and the sample was essentially liquid like at frequencies greater than 20 rad s−1 (Fig. 4A). The G′ and G″ of the 4∶1 mixture were 3000 ± 200 Pa and 400 ± 100 Pa, respectively, and the G′ and G″ of the 9∶1 mixture was 2900 ± 400 Pa and 320 ± 50 Pa. The G′ and G″ of both the 4∶1 and 9∶1 mixtures were an order of magnitude apart within error and were essentially independent of the applied frequency, consistent with the formation of rigid hydrogels.13 The gels of these co-fibrils had nearly identical rigidity to Fmoc–F5–Phe–OH gels. The response of the mixtures to 100% strain was tested as described above for Fmoc–F5–Phe–OH and Fmoc–F5–Phe–PEG (Fig. 4B). Interestingly both the 4∶1 and 9∶1 mixtures fully and rapidly (within 1 minute) recovered their original strength and maintained the order of magnitude difference between G′ and G″, unlike Fmoc–F5–Phe–OH-derived hydrogels (Fig. 4B). The 1∶1 mixture unexpectedly showed a significant increase in rigidity following the application and cessation of stress. After cessation of stress, the G′ and G″ increased to 950 ± 20 Pa and 22 ± 5 Pa, respectively, an ∼40-fold increase in rigidity. This increase in strength may be due to the forced entanglement of the PEG side chains on the fibrils from the oscillation of the rheometer head, imparting gel character. It should also be noted that while Fmoc–F5–Phe–OH-derived fibril networks precipitated upon application of stress, the co-assemblies with Fmoc–F5–Phe–PEG showed no evidence of precipitation, even after 4 weeks and repeated applications of stress.
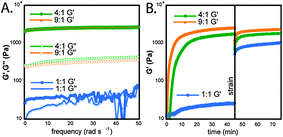 | ||
| Fig. 4 (A) Dynamic frequency sweeps of Fmoc–F5–Phe–OH∶Fmoc–F5–Phe–PEG co-fibrils (1∶1, blue; 4∶1, green; 9∶1, orange). (B) Dynamic time sweeps of each mixture before and after the application of 100% strain (G″ omitted for clarity). | ||
In conclusion, these results indicate that co-fibrils of Fmoc-F5–Phe–OH and Fmoc–F5–Phe–PEG exhibit the strong gel rigidity of Fmoc–F5–Phe–OH fibrils and the solvolytic stability of Fmoc–F5–Phe–PEG fibrils. The combination of these desirable attributes results in a hydrogel that exhibits ideal stress-responsive behaviors. These significant discoveries will enable the development of small molecule derived hydrogels that can be further functionalized for application to problems of biological significance.
We gratefully acknowledge Chris Willoughby and TA Instruments for use of an AR-G2 rheometer. This work was supported in part by DuPont (Young Professor Award to BLN), Alzheimer's Association (NIRG-08-90797), and the ACS PRF (48922-DNI).
Notes and references
- A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002–8018 CrossRef CAS.
- Y. Gao, Z. Yang, Y. Kuang, M. Ma, J. Li, F. Zhao and B. Xu, Biopolymers, 2010, 94, 19–31 CrossRef CAS.
- A. Mahler, M. Reches, M. Rechter, S. Cohen and E. Gazit, Adv. Mater., 2006, 18, 1365–1370 CrossRef CAS.
- R. Orbach, L. Adler-Abramovich, S. Zigerson, I. Mironi-Harpaz, D. Seliktar and E. Gazit, Biomacromolecules, 2009, 10, 2646–2651 CrossRef CAS.
- A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37–41 CrossRef.
- D. J. Adams, L. M. Mullen, M. Berta, L. Chen and W. J. Frith, Soft Matter, 2010, 6, 1971–1980 RSC.
- D. M. Ryan, S. B. Anderson, F. T. Senguen, R. E. Youngman and B. L. Nilsson, Soft Matter, 2010, 6, 475–479 RSC.
- D. M. Ryan, S. B. Anderson and B. L. Nilsson, Soft Matter, 2010, 6, 3220–3231 RSC.
- L. Haines-Butterick, K. Rajagopal, M. Branco, D. Salick, R. Rughani, M. Pilarz, M. S. Lamm, D. J. Pochan and J. P. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7791–7796 CrossRef CAS.
- O. D. Krishna and K. L. Kiick, Biopolymers, 2010, 94, 32–48 CrossRef CAS.
- T. S. Burkoth, T. L. S. Benzinger, D. N. M. Jones, K. Hallenga, S. C. Meredith and D. G. Lynn, J. Am. Chem. Soc., 1998, 120, 7655–7656 CrossRef CAS.
- J. H. Collier and P. B. Messersmith, Adv. Mater., 2004, 16, 907–910 CrossRef CAS.
- F. M. Menger and K. L. Caran, J. Am. Chem. Soc., 2000, 122, 11679–11691 CrossRef.
- L. E. Buerkle, Z. Li, A. M. Jamieson and S. J. Rowan, Langmuir, 2009, 25, 8833–8840 CrossRef CAS.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Materials and methods, synthesis details, digital images of gel, molecular model of fibrils, TEM images, UV-vis spectra, and fluorescence emission spectra. See DOI: 10.1039/c0cc02217a |
| This journal is © The Royal Society of Chemistry 2011 |