The mystery of gold's chemical activity: local bonding, morphology and reactivity of atomic oxygen
Thomas A.
Baker
b,
Xiaoying
Liu
a and
Cynthia M.
Friend
*ab
aDepartment of Chemistry and Chemical Biology, Harvard University, 12 Oxford St, Cambridge, MA 02138
bSchool of Engineering and Applied Sciences, Harvard University, Cambridge MA 02138. E-mail: cfriend@seas.harvard.edu; Tel: 01-617-495-4052
First published on 22nd November 2010
Abstract
Recently, gold has been intensely studied as a catalyst for key synthetic reactions. Gold is an attractive catalyst because, surprisingly, it is highly active and very selective for partial oxidation processes suggesting promise for energy-efficient “green” chemistry. The underlying origin of the high activity of Au is a controversial subject since metallic gold is commonly thought to be inert. Herein, we establish that one origin of the high activity for gold catalysis is the extremely reactive nature of atomic oxygen bound in 3-fold coordination sites on metallic gold. This is the predominant form of O at low concentrations on the surface, which is a strong indication that it is most relevant to catalytic conditions. Atomic oxygen bound to metallic Au in 3-fold sites has high activity for CO oxidation, oxidation of olefins, and oxidative transformations of alcohols and amines. Among the factors identified as important in Au–O interaction are the morphology of the surface, the local binding site of oxygen, and the degree of order of the oxygen overlayer. In this Perspective, we present an overview of both theory and experiments that identify the reactive forms of O and their associated charge density distributions and bond strengths. We also analyze and model the release of Au atoms induced by O binding to the surface. This rough surface also has the potential for O2 dissociation, which is a critical step if Au is to be activated catalytically. We further show the strong parallels between product distributions and reactivity for O-covered Au at low pressure (ultrahigh vacuum) and for nanoporous Au catalysts operating at atmospheric pressure as evidence that atomic O is the active species under working catalytic conditions when metallic Au is present. We briefly discuss the possible contributions of oxidants that may contain intact O–O bonds and of the Au–metal oxide support interface in Au catalysis. Finally, the challenges and future directions for fully understanding the activity of gold are considered.
 Thomas A. Baker | Thomas Baker received his Bachelor of Science degree in Chemistry from Indiana University of Pennsylvania and his PhD in Chemical Physics at Harvard University under the supervision of Professors Cynthia Friend and Efthimios Kaxiras. At Harvard, he used electronic structure calculations to study adsorption and reactions on gold surfaces. He is currently a post-doctoral researcher at UC Berkeley under the supervision of Professor Martin Head-Gordon working on the development and application of electronic structure theories for catalysts and molecular interactions. |
 Xiaoying Liu | Xiaoying Liu earned her Bachelor of Science degree in Chemistry and her Master of Science degree in Inorganic Chemistry from Fudan University, China. She completed her PhD in Physical Chemistry at Harvard University under the supervision of Professor Cynthia Friend. Her research included mechanistic studies of selective functionalization of olefins and oxidation of alcohols on single-crystal surfaces. She is currently a post-doctoral researcher at Massachusetts Institute of Technology under the supervision of Professor Klavs Jensen working on heterogeneous catalysis in continuous flow microreactor systems. |
 Cynthia M. Friend | Cynthia Friend, T. W. Richards Professor of Chemistry and Professor of Materials Science at Harvard University, focuses on the development of a molecular understanding of interfacial problems with the potential for high impact on critical problems in energy technology, heterogeneous catalysis, and growth of materials for device applications in her research. She combines studies of chemical reactions, atomic-scale imaging, spectroscopy and atomistic theory. Friend has been on the Harvard faculty since 1982 and was recently honored by election as a Fellow of the American Chemical Society (ACS) in 2010. |
1. Introduction
Over the centuries, gold has been valued for its luster and seeming inertness. For this reason, the discovery that Au is active for promoting a variety of molecular transformations stimulated considerable excitement and controversy in the scientific community. Remarkably, a wide range of oxidative transformations are promoted by gold at relatively low temperature and with very high selectivity. Thus, gold catalysts have great potential for energy-efficient “green” chemistry applications.1–3Gold-based materials with a wide range of size scales catalyze selective oxidative reactions. Nanoparticles of gold supported on metal oxides catalyze, for example, the oxidation of CH44,5 and CO,5,6water gas shift,7–9epoxidation of propene,10 selective oxidation of secondary alcohols,11 oxidative coupling of primary alcohols to esters,12,13 and acylation of amines.14 Micron-scale gold particles also promote transformations of amines, isocyanides and azo compounds in solution under aerobic conditions.15–19 Nanoporous Au, an unsupported high surface area material that contains ∼1–5% Ag, is also catalytically active for CO oxidation,20,21methanol esterification,22 and selective oxidation of glucose23 at atmospheric pressure and near room temperature. Clearly, gold in very different forms is highly active for a broad range of reactions and under various conditions—there is a vast literature on heterogeneous catalysis of gold, only some of which is cited here.
The origin of Au catalytic activity is still a topic of intense debate. There is a preponderance of evidence that bulk-like metallic Au—i.e. micron scale15–19,24–26 and nanoporous materials20–23—are effective and stable catalysts for several different types of reactions under aerobic conditions.15–19,24–26 On one hand, there is the view that nanoscale Au has special electronic properties that imparts to its chemical activity.6,27,28
In this Perspective article, we will focus on the role of atomic oxygen bound to metallic gold because it is clear from studies of bulk metallic Au crystals, micron-scale gold particles, and nanoporous Au that atomic oxygen bound to metallic gold is extremely active for oxidation reactions. Herein, we relate the structural and electronic properties of O on Au and of the Au itself to reactivity and selectivity in an effort to understand the underlying bonding factors that lead to high reactivity. We also briefly consider the possible role of molecular oxygen species and discuss future directions in research that will further unravel the mystery of gold activity for catalysis. Many of the issues raised in the discussion of Au activity apply broadly to understanding catalytic behavior in general; thus, we will generalize the discussion somewhat to the other coinage metals.
2. Overview of model studies on Au(111)
2.1 High activity of atomic oxygen on metallic gold
There is compelling evidence for the high activity of atomic oxygen bound to metallic Au for promoting heterogeneous oxidative transformation from fundamental studies on crystalline gold. The Au(111) surface has been used as a model in most cases; justified by the fact that it is the most thermodynamically stable phase and it has been identified as a predominant face for supported nanoparticles using high resolution electron microscopy.29The high activity of atomic oxygen bound to Au is established by model studies on single-crystal gold (Fig. 1). Atomic oxygen bound to single-crystal gold is highly active for CO oxidation30–34 and for activation of O–H, C–H, and N–H bonds in a variety of organic molecules.35–44 Many of the reactions on single-crystal gold parallel those observed on supported and unsupported Au at higher pressures and in solution when O2 is used as an oxidant as discussed above; thus, establishing the relevance of these studies.
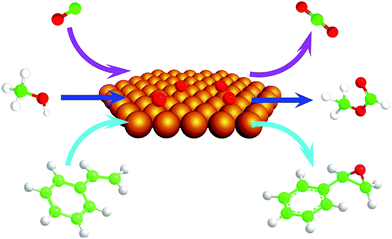 | ||
| Fig. 1 Scheme for reactions of CO (upper arrows), methanol (middle arrows), and styrene (lower arrows) with atomic oxygen deposited on a single-crystal gold surface to form CO2, methyl formate, and styrene oxide, respectively. | ||
Studies carried out under low pressure (ultrahigh vacuum conditions) have successfully modeled key catalytic processes by establishing molecular mechanisms for processes on coinage metals, most notably the selective oxidation of methanol to formaldehyde over Ag45,46 and, recently, the oxidative coupling of methanol over Au.22,39 Modeling the reactivity of coinage metals, especially Au, at low pressures is possible because of the relatively low reactivity of gold itself; thus, spectator species, including water, do not build up on the surface and there is a low steady-state coverage of oxygen even under higher pressure conditions due to the low dissociation probability of O2.
The low dissociation probability of O2 is most likely one of the primary factors limiting the rates of oxidative catalysis on Au and it is an impediment to investigating the nature of oxygen bonding on the surface. One of the explanations for the enhanced activity of supported Au nanoparticles is that there is a large fraction of edges containing under-coordinated Au atoms that are more efficient in dissociating O247,48 and that the residence time of molecules that are being oxidized is increased at these edge sites.47
The activity of O on metallic Au crystals fits a pattern of Brønsted acid–base reactions, similar to that described for Ag.49 Among the reactions promoted by O adsorbed on gold single crystals are olefin epoxidation,37,38,50propene oxidation to acrolein,40,43 selective oxidation of secondary alcohols,35,51 oxidative coupling of alcohols to esters,22,36,39,41,52oxidation of amines44 and ammonia, and oxidative coupling of aldehydes to secondary amines53 and alcohols36 to yield amides and esters, respectively. All of these processes require the presence of atomic oxygen on the Au surfaces—in the absence of O on the surface, there is no detectable reaction. These reactions are also extremely facile, in most cases occurring below room temperature.
The high activity of O on Au is clear, rendering it important to understand the bonding and reactivity of different configurations of O in order to further understand the activation of gold as a catalyst, even though the mechanisms for supplying O to Au are still a topic of discussion. It is particularly important to understand the local bonding of O on Au, the degree of interaction between neighboring oxygen atoms on the surface, and the morphology of the Au because they are all important factors in determining the activity and selectivity for reactions, including CO31oxidation and esterification of methanol39 and ethanol41 on O-covered Au(111).
The bonding configuration of O on Au is a key factor in the CO oxidation rate and in determining the selectivity for, e.g.olefin partial oxidation and alcohol esterification. The rate of CO oxidation, for example, strongly depends on the coverage and distribution of O on the surface, which is indicated by the variation in the rate of CO2 production by preparing the oxygen layer at two different surface temperatures (Fig. 2). Imaging and spectroscopic studies show that these two different preparation temperatures, for example, produce a surface with different local bonding of O and with different surface morphologies, as discussed below (Fig. 3). Both the activity and selectivity depend strongly on the O bonding environment for more complex reactions, such as styrene epoxidation37 and methanol esterification.39
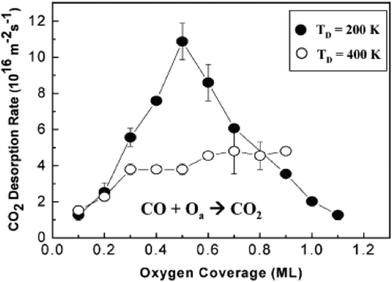 | ||
| Fig. 2 The rate of CO oxidation on Au(111) depends on the coverage of atomic oxygen and on the morphology and atomic order of the layer which is varied by using different temperatures to deposit the O. The black circles are for an O layer prepared by deposition of O using ozone at 200 K, which produces small Au particles containing O that is mainly bound in 3-fold sites. The open circles are for deposition of O using O3 dosing at 400 K, for which larger, ordered islands form a 2-dimensional oxide. Fig. 3 below shows STM images of these surfaces prior to reaction with CO. Data reprinted from ref. 31. | ||
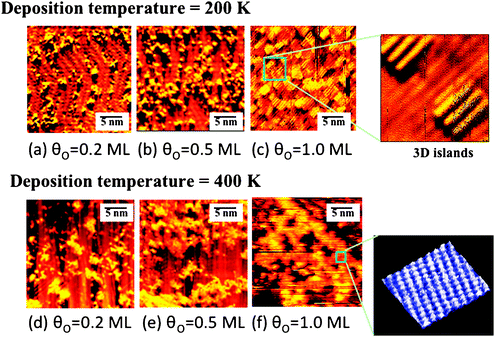 | ||
| Fig. 3 Scanning tunneling microscopic images showing the formation of nanoparticles on Au(111) after deposition of atomic O using ozone decomposition at two different surface temperatures. The top images are for deposition at 200 K for coverages of (a) 0.2 ML, (b) 0.5 ML, and (c) 1.0 ML. The bottom series of images are for deposition at 400 K for (d) 0.2 ML, (e) 0.5 ML, and (f) 1.0 ML. The insets show zoomed in images with atomic-level detail. The size of the islands increases with increasing O concentration and with the surface temperature. In the early stages of deposition, the herringbone reconstruction is still visible and islands appear to initially form at the elbow sites. Reprinted with permission from ref. 31. | ||
In model experimental studies, atomic oxygen is deposited on the surface and its reactivity investigated as a function of temperature and surface concentration. Corresponding theoretical studies similarly provide insight into the bonding of these oxygen species. Because the rate of O2 dissociation is essentially undetectable on flat, well-prepared gold surfaces, including Au(111), other methods for delivering oxygen to the surface must be used in order to investigate O bonding and reactivity. The methods that have been used to deliver oxygen atoms to Au surfaces are electron bombardment of condensed NO2,54 ion sputtering with O2,55,56 thermal dissociation of gaseous O2 using hot filaments,57 exposure to an O atom source,32 and decomposition of ozone.58 These methods all lead to the deposition of oxygen; however, there is substantial variation in surface morphology. In all cases, Au atoms are released from the surface upon oxygen adsorption, leading to complex surface structures.
Decomposition of ozone induces release of Au atoms from the surface to form particles that contain both Au and O, as discussed in detail below. Most reactivity studies in our lab were performed after depositing O on Au using O3. The release of Au atoms induced by O is also a potentially important pathway for sintering of supported Au nanoparticles, which is a major problem for catalyst deactivation. Therefore, the release of Au atoms due to O binding to the surface is an important issue to consider.
Electronic structure studies provide insight into the local bonding of oxygen on Au and into the release of Au from the surface. Theory is essential for a detailed interpretation of spectroscopic and imaging experiments and, in turn, understanding experimental trends in activity and selectivity for specific reactions. Theory provides insight into the types of oxygen species and structures that prevail under different conditions of temperature and surface concentration and how they relate to activity and selectivity. The combination of density functional theory calculations and experimental measurements have proven to be fruitful in understanding catalytic processes and even in designing new ones.59,60
2.2 Bonding configurations of O on Au(111) and Au atom release
The bonding of oxygen to gold surfaces induces morphological changes through the release of Au atoms from the surface, based on experimental studies (Fig. 3); thus, the theoretical treatment of O–Au bonding must consider the underlying causes for metal atom release and the effect of defects on O binding to the surface. The release of Au atoms is also induced by other electronegative species, sulfur61,62 and chlorine,63,64 suggesting that alteration of the charge density near the surface leads to the release of Au from the bulk. The O-induced release of Au from the surface is important because the morphology of the surface is correlated with the gold-oxygen interaction; affecting the type of oxygen species present and the corresponding reactivity of the surface. The mobilization of gold by oxygen is also a pathway for sintering of Au nanoparticles, which is a major technical impediment to practical implementation of supported Au catalysts. Our model experimental and theoretical studies provide key information on local O–Au bonding and on Au atom release.Release of Au atoms from the surface is induced by O binding, leading to the formation of particles that contain both O and Au. Scanning tunneling microscopy unequivocally demonstrates that the Au-containing particles are formed because the morphology remains essentially unchanged when all O is removed through CO oxidation at a surface temperature of 300 K or below (Fig. 4). The presence of O on these particles is inferred from the fact that larger, ordered particles formed at 400 K have a rectangular unit cell (Fig. 3f). If the particles were only Au alone, the unit cell would be hexagonal. The smaller particles formed at 200 K order and form larger islands with a rectangular unit cell when heating to 400 K; thus, confirming that the particles contain O and also that the small particles present at 200 K are kinetically-controlled structures.
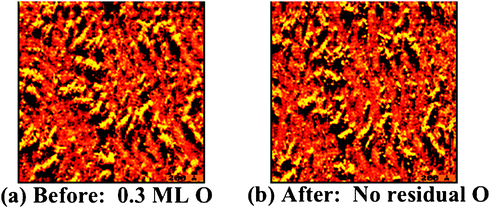 | ||
| Fig. 4 Scanning tunneling microscope images show that the Au surface morphology remains essentially unchanged after removal of all atomic O viaCO oxidation at 300 K; thus, the particles observed in the images contain Au. (a) A gold surface containing 0.3 ML of atomic O at 300 K; and (b) the same area of the surface observed with in situ scanning of the surface while reacting with CO (p = 5 × 10−8 torr for 5 min, 300 K) to remove all O. That the O was quantitatively removed by this treatment was confirmed in separate experiments using analogous treatment followed by temperature programmed experiments in which no O2 evolution at ∼550 K, characteristic of O recombination on Au, was detected. | ||
The roughness and the size distribution of the particles depend on the method used to deposit O and the surface temperature.65,66 Rapid rates of oxygen deposition, e.g. using ozone decomposition, lead to nucleation of small O-containing Au particles on the surface when the surface temperature is below 300 K. The nucleation of small particles is attributed to a short diffusion length before mobile O–Au species encounter each other, which leads to nucleation. The resulting morphology at 200 K for a surface containing ∼0.2 ML of O, has non-crystalline particles that are mostly on the order of 2 nm in diameter (Fig. 3a).31 This morphology is kinetically controlled, so that heating of the surface to above room temperature leads to an increase in the size and order of the O/Au particles, presumably through enhancement in diffusion (Fig. 3d). The particle size and degree of order of the O structure on the particles also increase as the total amount of O is increased on the surface. In contrast, slower deposition of O through electron-induced dissociation of nitrogen dioxide on Au(111), also leads to release of gold from the “elbows” in the herringbone structure; however, a smoother surface is formed with small islands and serrated step edges are observed due to the release of gold atoms from elbow sites of Au(111).67
Density functional theory (DFT) provides insight into the bonding of O on Au and on the release of Au atoms induced by surface-bound O.68 For low surface concentrations of O on Au(111) slabs, binding to sites with local three-fold coordination of O to Au is most energetically favorable—both with and without defects on the surface (Fig. 5). The effects of defects—adatoms, Au dimers, and Au vacancies—were explicitly studied in order to gain insight into bonding on defective surfaces from which Au atoms were released. In these studies, low O concentrations (1/16 of a monolayer; i.e. 1 O per 16 Au surface atoms in the repeating unit cell of the defect-free surface) were investigated. Oxygen bound in a 2-fold coordination site was found to be energetically less favorable by 0.49 eV. Our results are in general agreement with the groups that have also investigated oxygen adsorption on gold using DFT; however, the possibility of gold release from the surface induced by adsorbed O was not considered.69
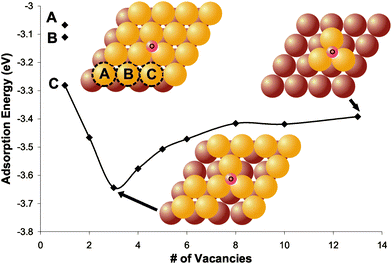 | ||
| Fig. 5 Density functional theory calculations show that the most stable binding configuration of O on a Au(111) slab is in a 3-fold coordination site—both with and without defects. Direct binding to defects is never preferred; however, defects, e.g.Au atom vacancies, stabilize O binding in neighboring sites. The adsorption energy of atomic oxygen as a function of the number of vacancies on Au(111), shown, illustrates the effect of defects. With only one vacancy there are three different configurations tested. The gold atom removed to create the vacancy is labeled (A, B, or C) on the model in the figure and its corresponding data point is also labeled. Models also show the atoms removed to create three vacancies and the final configuration with the maximum number of vacancies. Lighter yellow and darker brown large circles represent the top and second layer of gold, respectively. It is important to point out that this system serves as only a model; it is more appropriate to discuss the qualitative trends than the quantitative number of vacancies. Reprinted from ref. 68. | ||
Binding to sites of local three-fold coordination was always favored for this low oxygen concentration, even when bound in the vicinity of nearby defects. Direct binding to the defects was never favorable relative to the 3-fold site. The O–Au binding was destabilized by nearest neighbor Au adatoms and dimers; whereas, Au vacancies stabilized O bound in the neighboring three-coordination sites.
Atomic oxygen bound in local 3-fold coordination sites on rough, disordered surfaces are most active for oxidation of, e.g.CO (Fig. 2) and selective oxidation of styrene,37propene,40phenyl propenes,38,70 and alcohols.39,41 We use a combination of surface imaging, vibrational spectroscopy, and density functional theory to characterize the most active O species in our reactivity studies.
The identification of O bound in 3-fold coordination sites as the predominant species at low oxygen concentrations is validated by comparing computed to experimentally-measured vibrational frequencies. At low O coverages, there is a single, broad vibrational peak at ∼360 cm−1 measured using electron scattering (Fig. 6). Computed frequencies are in the same range, with some variation in the vibrational frequency depending on proximity to various types of defects (Table 1). The computed spread in energy is within the envelope of measured frequencies, indicating that the breadth of the peak is due to heterogeneity of the surface and the intrinsic limitations of the resolution of the measurements.
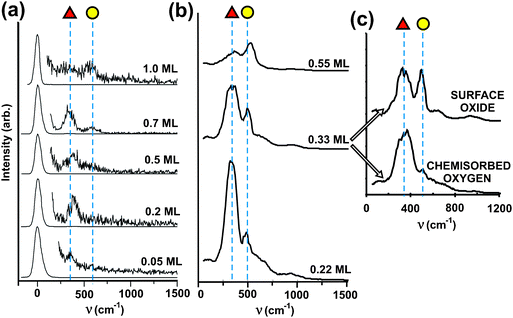 | ||
| Fig. 6 Experimental and simulated vibrational spectra for O bound to Au(111) with varying O surface concentration. (a) High resolution electron energy loss (HREEL) data obtained after depositing O onto Au(111) using ozone decomposition at 200 K. All spectra are on the same scale, with intensities multiplied by a factor of 100. Two main peaks are identified with dotted lines at 360 (red triangle) and 570 cm−1 (yellow circle). (b) Calculated vibrational spectra for different coverages of atomic oxygen introduced to the surface in AIMD simulations described below. The red triangle labels the peak at 350 cm−1 and the yellow circle one at 500 cm−1. The spectrum simulated for an O coverage of 0.33 ML split in (c) into MD runs that produced chemisorbed oxygen in 3-fold sites (red triangle) and oxygen in a “surface oxide”, which has two O atoms bound to a single Au atom, which is pulled out of the surface. Reprinted with permission from ref. 71. | ||
| System | v (cm−1) |
|---|---|
| FCC | 405.5 |
| Top of single adatom | 674.9 |
| FCC on 2D island | 421.9 |
| Edge of step | 452.5 |
| Next to one vacancy | 370.3 |
| Bridge on 2 Au adatoms | 491.9 |
| FCC on Au strained 92% | 442.5 |
| FCC on Au strained 108% | 405.9 |
The stabilization of Au–O bonding via interaction with nearby vacancies is consistent with the enhanced thermal stability of sputtered surfaces containing O relative to clean surfaces sputtered to have the same rough morphology. Hence, it is not direct bonding to defects that leads to this stabilization; rather, interaction with neighboring vacancies increases Au–O bonding to the 3-fold sites.
2.3 Release of gold atoms and higher oxygen coverages
The release of Au atoms from the surface and incorporation of gold atoms in the adsorbate layer depends on the concentration of O on the surface, according to DFT studies. At high concentrations (coverages) of O, gold atom release from even a (111) slab is energetically favorable; at low coverages, coordination to the flat (111) surface is favored.69 The bonding between the O atoms and the gold substrate is also less ionic at higher O coverage, evaluated based on the smaller degree of charge transfer to the oxygen atoms and the increase of electron density in the region between the O and gold surface atoms.Two factors that contribute to gold incorporation and the roughened gold surface are identified through our calculations: (1) the adsorbate-gold bond becomes stronger and changes (more covalent-like with a smaller amount of charge transfer) when gold is incorporated and, (2) adsorbates can stabilize the presence of adatoms or other defects in the gold substrate. The first factor is not surprising since adsorption is often stronger with the presence of defects, and has been energetically verified for these systems in our previous calculations.71 However, stronger adsorption does not ensure that the structure that incorporates Au is the lowest in energy since there is an energetic cost for creating under-coordinated gold atoms on the surface.
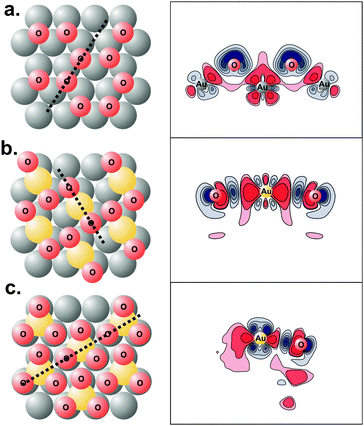 | ||
| Fig. 7 Lowest energy structures (left panels) and charge density difference plots (right panels) of: (a) 0.67 ML O adsorption on flat, (1 × 1) Au(111) surface; (b) 0.67 ML O adsorption on Au(111) surface covered with 0.33 ML adatoms; (c) 1.00 ML O adsorption on 0.33 ML Au adatom-covered Au(111) surface. The thick dotted lines on the structural figures on the left show the planes on which the density difference is plotted. Red contours correspond to charge depletion and blue contours to charge accumulation. Notice the localization of charge when oxygen is bound to gold adatoms. Reprinted with permission from ref. 69. | ||
Electronic density plots also show that the bonding of O on a surface containing gold adatoms is more covalent compared to O bound to a flat (111) slab, since bonding is more localized, Fig. 7.71 The stronger covalent interaction resulted in a smaller degree of charge transfer from gold to the adsorbate, imparting a smaller partial negative charge compared to binding to the flat Au(111). This result provides an explanation for the dependence of gold incorporation on the adsorbate coverage: as the coverage increases, the inter-adsorbate distance decreases and the Coulombic-repulsive interaction between the partially negatively charged adsorbates increases. Gold incorporation, however, decreases this negative charge since the adsorbate-gold bond becomes more covalent in nature. Hence, the repulsive interaction between adsorbates decreases.
There are several different types of oxygen bound to gold identified experimentally using spectroscopy and imaging studies, whose populations depend on the surface concentration of O and on the temperature of the surface (Fig. 3 and 6). There are clearly different types of O bound to Au that vary in concentration depending on coverage because there are two distinct vibrational peaks that vary independently with O coverage and adsorption temperature (Fig. 6a). Theory is necessary to associate specific bonding configurations with the spectroscopic and imaging results. Only one of these peaks is identified by the slab calculations at low O coverage, described above. Because different bonding configurations generally should lead to different chemical behavior, it is crucial to understand the local bonding of the various oxygen species and to obtain insight into what conditions favor specific structures.
We implemented ab initio molecular dynamics (AIMD) simulations to model the dynamic restructuring of the Au(111) surface due to the adsorption of atomic oxygen. Although DFT is a well-established tool for describing the bonding of atoms on surfaces using static periodic slab calculations, they require selection of a specific geometric configuration and unit cell. Oxygen atom binding to Au is complex and several different types of oxygen coexist on the surface with their populations depending on the surface temperature and concentration of O on the surface. Hence, except at low coverage, where there is a single, predominant bonding configuration identified using DFT and vibrational spectroscopy as O in sites of local 3-fold coordination, static calculations could not be used to study the other types of O; instead, AIMD simulations were used.71
Three different types of O binding to Au were clearly identified in simulations performed for three different O coverages (0.22 ML, 0.33 ML, and 0.55 ML) and three different temperatures (200 K, 500 K, 800 K) using AIMD. We found that the morphology and local binding site of oxygen depend on the coverage and the surface temperature, in agreement with experimental results. Calculated vibrational frequencies derived from these simulations are in agreement with vibrational spectroscopy experiments (Fig. 6).31
Three different types of O were identified in our simulations (Fig. 8): (1) chemisorbed oxygen bound to a 3-fold hollow site, in agreement with the static DFT calculations at low coverage; (2) surface oxide, which incorporates gold into the structure; and, (3) subsurface oxide, in which O migrates below the surface, displacing Au outward. At low oxygen coverage (<0.33 ML) or temperature (200 K), the simulated Au(111) surface is smooth and oxygen on the surface is primarily chemisorbed in 3-fold coordination sites, in agreement with the predominance of the peak at 360 cm−1 measured experimentally. At higher coverages (> 0.33 ML) or temperatures (500 K and 800 K), a significant amount of surface and subsurface oxide species form, which have vibrational signatures at 500 cm−1.
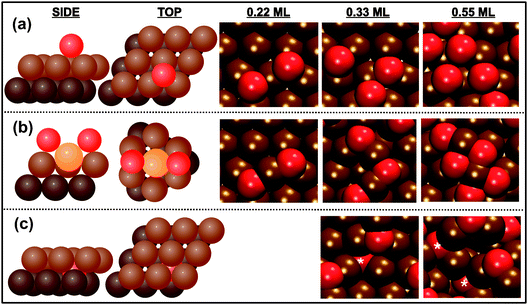 | ||
| Fig. 8 Three different types of O bonding to Au(111), shown schematically on the left, are identified using ab initio molecular dynamics simulations: (a) chemisorbed, (b) surface oxide, and (c) subsurface oxide. Dark brown, light brown, yellow, and red spheres represent the second layer of gold, top layer of gold, gold adatoms, and oxygen atoms, respectively. Illustrative examples from a specific trajectory are shown at the right. Brown and red spheres represent gold and oxygen atoms, respectively. A white asterisk labels subsurface oxygen atoms in (c). The subsurface oxide was only found for oxygen coverages above 0.33 ML. Reprinted with permission from ref. 71. | ||
Our calculations suggest that gold adatom incorporation is energetically possible from both a thermodynamic and kinetic standpoint. In the static DFT calculations, we are necessarily calculating the thermodynamic structure, which indicate that gold incorporation is favorable at higher oxygen coverages. The AIMD simulations indicate that gold atom release is kinetically favored, since upon dosing oxygen, gold incorporation occurs on a very short timescale (typically within the first couple picoseconds). The AIMD simulations also indicate these structures are at least metastable, since they typically persisted for the duration of the simulation.
2.4 High activity of disordered chemisorbed O for CO oxidation
The reactivity of atomic oxygen clearly depends on local bonding of oxygen present on the gold surface as established by the combination of theory and experiment. Disordered oxygen bound in local 3-fold coordination sites formed at low temperatures and low coverages is most active for a wide range of reactions. This is clearly shown in the case of CO oxidation where the rate of CO oxidation is faster for the oxygen species that is predominant at low coverages and temperatures.72AIMD simulations of CO reaction with O on Au(111) also show that oxygen bound in 3-fold coordination sites is the most reactive. The adsorption of CO and its oxidation to CO2 on oxygen-covered Au(111) at 500 K were simulated for three different initial oxygen coverages: 0.22 ML, 0.33 ML, and 0.55 ML.73 The frequency of adsorption vs. reaction depends on the initial coverage of oxygen. Compared to adsorption on the oxygen-free surface, CO has a longer average residence time on the surface, however, the adsorption lifetime decreases with increasing oxygen coverage.
We have found the most reactive surface to be the one with 0.22 ML of oxygen, where O in 3-fold sites is the predominant species, and confirmed that this form of adsorbed O is most likely to react with incoming CO. This disagrees with the most reactive coverage found experimentally, because of the different coverage definitions and approximations in the computational model.73 The type of oxygen present before reaction is determined from separate AIMD calculations and the oxygen type that participates in each reaction is found by analyzing each independent AIMD trajectory that results in CO oxidation. The oxygen chemisorbed in 3-fold sites is involved in the majority of the oxidation reactions.
For 0.22 ML of oxygen coverage, initially 80% of the surface is covered with chemisorbed oxygen while 20% is covered in surface oxide, yet 86% of the CO oxidation events are with chemisorbed oxygen in 3-fold sites. Similarly, 83% of the reactive atoms are those in 3-fold sites, whereas they only comprise 60% of the total number of oxygen atoms in the layer initially containing 0.33 ML of oxygen. Even when the rate is normalized to take into account the ratio of each oxygen species on the surface the relative rate for reaction with chemisorbed oxygen in 3-fold sites is significantly higher than surface oxide (gold incorporation). We attribute the reactivity differences to the type of oxygen–Au bond (described in section 2.3) and to differences in the binding properties of CO, both which affects the transition state and reaction pathway for CO oxidation; rather, merely to changes in the binding strength of oxygen. While these experiments and corresponding work focused on CO oxidation, similar trends in reactivity and selectivity has been observed for other oxidation reactions on gold suggesting that chemisorbed oxygen formed at low temperatures and oxygen coverages is key to the reactivity of gold.
2.5 Interface and charging effects
Although we clearly demonstrate the activity of atomic O on metallic Au for a variety of oxidation reactions, there is also considerable evidence in the literature that metal oxide supports of the Au catalysts play an important role in determining their activity. In addition, electronic effects associated with small particles,74,75Au bilayers,76 charge transfer to or from Au,77 and dissolution of Au into the oxide lattice have all been discussed as factors in determining the activity of supported Au.There has also been considerable discussion about the role of the interface between the metal and support for oxidation. The interface has been proposed to provide sites for binding and transformation of reactants. Possible migration of active adsorbed species—especially oxygen—across the Au–metal oxide support has also been proposed. While there are some systems where the interface seems to not play an important role, in other cases, it is clear that interfaces or supports play a vital role. For example, the clean Au(111) surface is not catalytically active for the water gas shift reaction, but is very active when using an inverse catalyst where ∼30% of the gold is covered by ceria or titania nanoparticles.78,79 It is proposed that CO adsorbs on gold while water dissociation occurs at the interface. Another example of the important role of the interface is for glucose oxidation on gold nanoparticles. Gold nanoparticles supported on carbon materials or polymers showed no catalytic activity but substantial activity when supported on reducible metal oxides such as ZrO2, TiO2, and CeO2.43 In addition, the support can even affect the growth of gold nanoparticles on the surface.80,81
The oxidation state on supported gold is also proposed to have an impact on the activity. A model study of gold nanoparticles and adatoms on the (111) surface of single-crystal magnetite suggested that nonmetallic, positively charged Au species may play a key role in reactions involving CO. For gold supported on an oxide, the interaction between Au and the support alters the electronic structure of Au nanoclusters. Depending on the size of gold nanoparticle and the support, it is believed that both the oxidized and reduced states of gold can exist and the vibrational frequency of CO is used as a gauge of the charge state of Au.82–84 In particular, defects on the oxide support are thought to play a key role in anchoring the Au particles and in transferring electronic charge to Au, contributing to the special catalytic activity.85
While our studies do not address these various effects, none of these proposals negate the importance of atomic oxygen bound to Au as being active for a range of oxidation reactions. Additional study is required to more fully resolve these issues using a combination of experiment and theory.
2.6 Possible role of molecular oxygen
One of the primary issues discussed in the literature is how atomic O is supplied to gold with sufficient efficiency for catalytic activity, because it is largely agreed that the rate of O2 dissociation on flat gold is extremely low—essentially undetectable. Generally speaking, the discussion falls into two categories: (1) unusual electronic properties of small-scale Au, and (2) the effect of the Au–metal oxide interface. Electronic effects include the explanation that the dissociation of O2 is enhanced at coordinatively unsaturated sites on rough surfaces or Au particles which is supported by theoretical studies. The view that Au nanoparticles or Au bilayers have different electronic properties than the metallic form has been advanced. There is also discussion about the possible contribution of charged Au species—both cationic and anionic—in activating bonds in reactant species.77 Interface effects may also be electronic in origin in that the oxide may alter the electronic structure of the Au, leading to chemical activity.86–90 Reactive species, including atomic oxygen, migrate across the interface between a Au particle and the oxide support is another possible mechanism. Migration across the interface would be more efficient for small particles because of the larger interface length relative to the overall surface area.While experimental studies discussed herein clearly show that atomic O bound to Au is highly active for a wide variety of oxidative reactions, there is still a controversy about the types of oxygen that react with CO. Atomic O is clearly very active for CO oxidation; however, O2 dissociation is required under reaction conditions and it is rather inefficient. In contrast to other transition-metal surfaces, there is no appreciable O2 dissociation on extended single crystals of Au.91,92 Edge sites have been proposed to enhance O2 dissociation in nanoparticles;93,94 nevertheless, the rate of supply of O to the surface is relatively slow compared to other metal surfaces.95 Even though it is clear that atomic O is highly active for oxidative transformations on Au, the low dissociation probability raises the possibility of other reactive species.
An alternative reaction scenario involves molecular O2 adsorption followed by reaction with incoming molecules. For example, CO has been proposed to form a peroxo-like, OC⋯O2 complex, which leads to CO2 and residual atomic oxygen. Several theoretical and experimental studies suggest that O2 is involved in oxidative chemistry on Au particle.86,96
Mechanisms involving reaction of molecular oxygen on gold also suffer from the fact that the steady-state concentration at reaction temperatures will be extremely low because of the very weak bonding of O2 to gold. Experimentally, O2 binding to Au(111) is extremely weak, desorbing at very low temperature, ∼45 K.97 Density functional calculations likewise indicate that there is no binding of O2 to Au(111) and only weak binding to step edges of Au(211).47 The extremely weak binding is indicative of a minimal amount of charge transfer between molecular oxygen and the metal. This is in contrast to molecular oxygen adsorption on silver, which is much stronger,98 and for which there is evidence of a significant amount of charge transfer from the metal to the molecular O2 species: spectroscopic measurements indicated that ∼1.7 electrons are transferred to the adsorbed O2 complex through backbonding into the π* orbitals.99–103
The weak binding of O2 to gold renders it more difficult to study from a reactive chemistry point of view because a significant quantity cannot be maintained on the surface under low pressure conditions amenable to fundamental studies. Analogous studies of CO oxidation by O2 on Ag have been performed and CO2 production is observed; however, O2 is more strongly bound on Ag because of the significant amount of charge transfer.
There is also considerable evidence that peroxo species promote oxidation reactions on Au and Au-based alloy particles. The use of peroxides and related species also warrants further fundamental study. Our work has mainly focused on identifying the most reactive atomic oxygen species, but more work is needed both experimentally and theoretically to explore the possibility of other oxygen species participating in oxidation.
2.7 Comparisons to oxidation on silver surfaces
For many oxidation reactions, there are direct parallels and patterns between reactions on gold and silver.104 One of the reasons for the similarities is the fact that oxygen can act as a Brønsted base on both surfaces, thus can abstract hydrogen from olefins on the surface. However, due to the slight differences in the metal electronegativity values, we can expect a slightly different oxygen–metal interaction. Indeed, DFT studies found a large negative charge of similar magnitude for oxygen adsorbed on Cu(111) and Ag(111), whereas oxygen on Au(111) is significantly less ionic.105 This would seem to suggest that oxygen would not be as strong of a base on gold compared to silver, which has important implications for a wide range of reactions.For many reactions, the products and mechanisms are nearly the same on gold and silver. For example, the oxidation of styrene is very similar on Au(111)50 compared to that on Ag(111).106 But for other reactions, there are some important differences. The reaction of propene on silver results in undesired combustion, via an unstable allyl species that is formed through allylic hydrogen abstraction.78,79 This allyl species is formed because oxygen is a strong Brønsted base on the silver surface. On the other hand, on Au(111) a variety of partial oxidation products are observed, including acrolein, acrylic acid, and carbon suboxide, which are produced in competition with combustion.43 These partial oxidation products are produced through an allyloxy intermediate that is formed viainsertion of oxygen into the allylic C–H bond (a competing pathway to the allyl formation). The presence of this pathway on gold suggests oxygen is not as strong of a base on the gold surface compared to silver.
Differences between silver and gold are also observed for the oxidation of cyclohexene. On silver, benzene is the primary partial oxidation product, which is believed to be formed by the sequential abstraction of four hydrogen atoms around the ring. But on gold, additional oxidation products are formed, including 2-cyclohexen-1-one, 2-cyclohexene-1, 4-dione, and phenol.104 So while hydrogen abstraction still occurs on the gold surface, it seems that it is not as facile as on the silver surface and slow enough on gold to allow competing pathways that involve oxygen insertion. It is not within the scope of this perspective to completely discuss all the differences between the two metals, but there is an excellent review that outlines a much larger list of similarities and differences.104
3. Future outlook
The two main impediments for implementing Au catalysts in practical processes are the long-term stability of the catalysts and the rates of reaction. For mechanisms involving reactions of atomic oxygen on Au, the rate of supply must be increased, preferably via enhanced dissociation of O2. Alloys have already shown promise for increasing the rate of oxidation; for example, Au–Pd107 and Au–Ag22,108–110 alloys. Both fundamental experiment and theory will potentially play a role in understanding these alloys and understanding more complex heterostructures involving Au and metal oxide nanostructures.While we mainly discuss the reactivity of atomic oxygen in this Perspective, investigations of the possible role of O2 would provide further insight into the origin of gold’s catalytic activity under practical conditions. An example would be cryogenic trapping of O2 on Au followed by introduction of reactants, e.g.alcohols. Another possibility is to “trap” molecular O2 species using alloys, since O2 binds to other metals, e.g.Ag,111 more robustly than Au. On the other hand, the isolation of peroxide species for investigation would be even more difficult—it requires the removal of hydrogen from H2O2 on supported Au catalysts, not to mention the introduction of pure gaseous hydrogen peroxide onto the surface.
When considering the effects of the oxide support, fundamental surface science studies have the potential to provide much needed insight. For example, fundamental studies have been instrumental in understanding the strong metal–support interaction (SMSI) effect. There were several proposed models to explain the effect, including, (i) alloy formation (ii) electronic metal–support interactions and (iii) encapsulation, but it was not entirely clear which was correct.112 Recent fundamental work by the Bowker group113,114 has defined this effect more clearly, showing that encapsulation occurs. Similar types of studies could be applied to improve our understanding of support effects.
Fundamental studies that combine surface imaging, spectroscopy and reactivity have the potential to provide insight into the local bonding and redistribution of surface species during reaction that are important in determining product distributions. It is already clear from our studies of Au, for example, that the surface is incredibly dynamic under reaction conditions. Recent development of in situ methods for imaging and for X-ray photoelectron spectroscopy hold out the possibility to understand the dynamics of oxygen species as a function of temperature and the evolution of the surface reconstruction induced by adsorbates and reactive intermediates. Scanning tunneling microscopy (STM), in particular, is able to image processes that happen locally on surfaces, in contrast to the statistical behavior over many molecules or atoms obtained from other techniques. Furthermore, it can contribute to understanding of local chemical environments during the adsorption and reaction processes that may escape detection by other surface analytical techniques. STM images have been used to investigate the details of chemical reactions on surfaces, such as site specificity and monitoring the atomic and molecular dynamics.115,116
With the development of high-resolution and fast-scanning STM, it is possible to track closely the oxidation reaction promoted by surface oxygen in real space and real time with atomic resolution, and ultimately the identification of elementary steps. Further, STM allows in situ scanning of local structural change upon the introduction of one or more reactants, which can provide rich information on the evolution of reactive sites and the interaction between oxygen and the reactant. Importantly, STM, unlike most surface science tools, can operate under ambient conditions. Therefore, it holds great potential to achieve in situ investigation at high pressures in the range where most catalytic reactions are operated. STM can therefore be expected to play an important role in understanding the structure of oxygen species on gold, its dynamics as a function of temperature, and the dynamic evolution of the surface reconstruction induced by adsorbates and reactive intermediates.
It is important to note that the combination of STM and theory, such as DFT calculations, allows for elucidation of more quantitative information on the local electronic structure by matching simulated STM images with experimental measurement. Therefore it affords further understanding of the interaction type, bond strength, electron transfer process, and energetics of different reaction pathways.
The development and application of new theoretical methods to investigate the gold–oxygen interaction will undoubtedly provide valuable information and insight that is not currently available using experimental techniques. Furthermore, the development of these tools is not just useful for the gold–oxygen interaction, but also applies to a wide range of other systems. There are longstanding weaknesses of current DFT calculations and other theoretical tools that are exposed when modeling the Au/O system because of the robustness and complexity of the interaction.
A fundamental problem with describing the Au/O interaction is the fact that oxygen adsorption produces a roughened disordered gold surface. Plane-wave DFT calculations naturally lend themselves to systems that can be partitioned into small periodic unit cells; therefore, it is difficult to model the non-periodic disordered gold surface by this approach. One practical solution to this problem is to employ a large unit cell. DFT or other ab initio calculations are difficult to implement because of their computational cost (DFT typically scales as ∼N3 where N is the number of the electrons in the system, other more accurate methods which better model electron correlation, like coupled-cluster, perturbative treatments, or configuration interaction, scale at much higher costs). Less accurate methods, like classical molecular mechanics or tight binding, are much faster, but are not generally suited for metal surfaces. Future theoretical developments will need to either reduce the computational cost so that larger unit cells can be modeled or develop clever methods to capture the ‘long length-scale’ disorder with smaller unit cells. The success of our AIMD calculations suggest that by including the dynamics of the system we can partially model the disordered surface, or at least capture the important local interactions so as to accurately reproduce experimental vibrational spectra.
Ideally, detailed theoretical treatment of more complex oxidative reactions on gold would also be possible. Either computational observation of reactions via modeling of dynamics, or searching for transition states and their barriers using static, periodic calculations can be envisioned. Modeling the dynamics of a reaction, including the effects of temperature, is a major challenge for theoretical studies. Kinetic Monte Carlo (kMC) techniques have been successfully applied to some problems; for example, CO oxidation on RuO2(110).117 Unfortunately, kMC methods require a knowledge of all events important to the dynamics of the system. Furthermore, the spatial degrees of freedom of the system are typically reduced to a simple lattice. The interaction of oxygen with Au is complex because of the role of defects and metal atom release and further reaction with an olefin is likely to involve these defects and many complicated and unforeseen reaction pathways. On the other hand, molecular dynamics requires calculation of the forces between atoms in the system. Classical force fields (molecular mechanics) are computationally cheap, so that relative large systems can be treated—thousands of atoms can be simulated for nanoseconds.118 Unfortunately, these force fields are not very accurate and often do not capture complex chemical behavior, including bond breaking and formation. Ab initio molecular dynamics (AIMD) is more accurate; however, it is computationally expensive and, therefore, is restricted to short time scales. Furthermore, AIMD employs the Born–Oppenheimer approximation so that electronic non-adiabatic coupling, which may be important in catalytic reactions, cannot be treated. There are only a few cases, e.g.CO oxidation on oxygen-covered Au(111) that occur on short enough time scales to be successfully modeled using AIMD. Clearly, methods are needed for enhancing the speed of computation for molecular dynamics simulations. This is a major challenge for future studies.
The second broad method for understanding a reaction on a surface is to map out the potential energy surface and to compute barrier heights for specific elementary steps. Using transition state theory, barrier heights and pre-exponential factors can be used to calculate the rate of a reaction. When calculating the transition state structure, a highly accurate energy is needed, meaning that in most cases a quantum-mechanical treatment is necessary. But even the relatively accurate quantum mechanical treatments can provide inaccurate energies for transition states. Modern DFT can provide accurate estimates of, for example, lattice constants, bond distances, and adsorption energies for a wide range of metals and other solids with the GGA functional.119 However, there have been many problems with modeling transition states, mainly due to the well-known self-interaction problem, thus resulting in inaccurate energies and thus reaction rates.
If the potential energy surface or reaction space is complex, transition state theory is difficult to apply. On a complex surface e.g. the disordered O/Au(111) surface, there are a wide variety or ensemble of possible pathways that depend on the local oxygen binding, the location and orientation of other reactants, and the configuration of metal atoms. When using transition state theory to understand a reaction on a surface, all of these pathways (or at least the ones that are lowest in energy) must be taken into account; however, important pathways may be missed because they were not explicitly considered.
The contribution of entropy to the free energy is also an important factor in modeling reaction kinetics on surfaces. Changes in entropy (or the entropy of activation) can be approximated using DFT by calculating the partition function, which can be found within the harmonic approximation by calculating vibrational frequencies. Furthermore, the temperature dependence of enthalpy is often not considered. The entropic contributions in some catalytic systems may be small enough (<0.1 eV) to be neglected, but there are likely systems where entropic contributions and zero-point energy corrections can have a significant contribution. Valdes et al. found these contributions to be ∼0.4 eV for reactions important for the oxidation and photo-oxidation of water on rutile TiO2(110).
Overall, there are advances required to more effectively understand and model catalytic processes, especially for complex reactions, such as olefin oxidation or alcohol coupling, where selectivity is a concern. There is a continued need for close connection between theory and experiment in order to advance our understanding of complex reactions important in heterogeneous catalysis.
4. Conclusions
Atomic oxygen bound to metallic Au activates a variety of chemical bonds that are important for catalytic oxidation reactions in a predictable pattern of reactivity that strongly parallels processes under catalytic operating conditions; thus, providing strong evidence that atomic O on metallic Au is the active oxidant in catalytic processes. Experimental observations and theoretical calculations show that oxygen atoms bound to the surface of gold induces release of Au atoms, leading to formation of a rough, disordered surface that has the potential for O2 dissociation, but also forms an extremely active O species that is bound to sites of local three-fold coordination. Atomic oxygen chemisorbed in these 3-fold coordination sites is most active for CO oxidation—both experimentally and in molecular dynamics simulations. It is also the predominant species at low O surface concentrations, which are most relevant to catalytic conditions. Low coverages of O also favor higher activity and higher selectivity in oxidative transformations of olefins and alcohols in experimental studies.Two other types of O binding to Au are identified in our work: an ordered surface oxide in which two O atoms are bound to a single Au atom and a subsurface oxide with oxygen bound below the surface plane. These two types of O are less reactive for CO oxidation—based on both experiment and theory. They are also less active and less selective for olefin oxidation and oxidation of alcohols on Au, based on experimental observations. These types of O are formed at higher coverages and also higher temperatures.
We also show that Au atoms are very labile, even on extended surfaces, and that binding of O to Au induces release of Au atoms. Two factors that contribute to gold incorporation and the roughened gold surface are identified through our calculations: (1) the adsorbate-gold bond becomes stronger and changes (more covalent-like with a smaller amount of charge transfer) when gold is incorporated and, (2) adsorbates can stabilize the presence of adatoms or other defects in the gold substrate. The release of Au atoms is important in determining reactivity, but also provides a pathway for the undesirable sintering of Au nanoparticles. Hence, materials design to kinetically impede sintering is needed for practical realization of Au catalysis.
The interaction of oxygen with gold is an important system to understand in its own right and it underscores the need for including complexity in theoretical models of heterogeneous reactions. With the combination of experiment and theory, fundamental knowledge has been developed on the local bonding properties of oxygen species with Au, the correlation between surface morphology and local coordination, the intrinsic characteristics of active oxygen species, and the reactivity for CO oxidation. By combining experiment with theory, we can expect more exciting discoveries in the area of atomic- and molecular-scale structure and dynamic evolution of active sites, surface morphology/reconstruction, and interactions between oxygen and adsorbates during oxidation reactions.
Acknowledgements
The authors gratefully acknowledge support of this work by the U.S. Department of Energy, Basic Energy Sciences, under grant no. DE-FG02-ER84-13289.References
- N. Dimitratos, J. A. Lopez-Sanchez and G. J. Hutchings, Top. Catal., 2009, 52, 258–268 CrossRef CAS.
- B. K. Min and C. M. Friend, Chem. Rev., 2007, 107, 2709–2724 CrossRef CAS.
- L. Prati, P. Spontoni and A. Gaiassi, Top. Catal., 2009, 52, 288–296 CrossRef CAS.
- R. J. H. Grisel, P. J. Kooyman and B. E. Nieuwenhuys, J. Catal., 2000, 191, 430–437 CrossRef CAS.
- R. J. H. Grisel and B. E. Neiuwenhuys, Catal. Today, 2001, 64, 69–81 CrossRef CAS.
- M. Haruta and M. Date, Appl. Catal., A, 2001, 222, 427–437 CrossRef CAS.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935–938 CrossRef CAS.
- Q. Fu, A. Weber and M. Flytzani-Stephanopoulos, Catal. Lett., 2001, 77, 87–95 CrossRef CAS.
- N. Yi, R. Si, H. Saltsburg and M. Flytzani-Stephanopoulos, Energy Environ. Sci., 2010, 3, 831–837 RSC.
- T. Hayashi, K. Tanaka and M. Haruta, J. Catal., 1998, 178, 566–575 CrossRef CAS.
- H. Sakurai, I. Kamiya, H. Kitahara, H. Tsunoyama and T. Tsukuda, Synlett, 2009, 245–248 CrossRef CAS.
- B. Jorgensen, S. E. Christiansen, M. L. D. Thomsen and C. H. Christensen, J. Catal., 2007, 251, 332–337 CrossRef CAS.
- I. S. Nielsen, E. Taarning, K. Egeblad, R. Madsen and C. H. Christensen, Catal. Lett., 2007, 116, 35–40 CrossRef CAS.
- S. K. Klitgaard, K. Egeblad, U. V. Mentzel, A. G. Popov, T. Jensen, E. Taarning, I. S. Nielsen and C. H. Christensen, Green Chem., 2008, 10, 419–423 RSC.
- B. L. Zhu, M. Lazar, B. G. Trewyn and R. J. Angelici, J. Catal., 2008, 260, 1–6 CrossRef CAS.
- B. L. Zhu and R. J. Angelici, J. Am. Chem. Soc., 2006, 128, 14460–14461 CrossRef CAS.
- Y. B. Zhou, B. G. Trewyn, R. J. Angelici and L. K. Woo, J. Am. Chem. Soc., 2009, 131, 11734–11743 CrossRef CAS.
- Y. B. Zhou, R. J. Angelici and L. K. Woo, Catal. Lett., 2010, 137, 8–15 CrossRef CAS.
- R. J. Angelici and M. Lazar, Inorg. Chem., 2008, 47, 9155–9165 CrossRef CAS.
- V. Zielasek, B. Jurgens, C. Schulz, J. Biener, M. M. Biener, A. V. Hamza and M. Baumer, Angew. Chem., Int. Ed., 2006, 45, 8241–8244 CrossRef CAS.
- C. Xu, X. Xu, J. Su and Y. Ding, J. Catal., 2007, 252, 243–248 CrossRef CAS.
- A. Wittstock, V. Zielasek, J. Biener, C. M. Friend and M. Baumer, Science, 2010, 327, 319–322 CrossRef CAS.
- H. M. Yin, C. Q. Zhou, C. X. Xu, P. P. Liu, X. H. Xu and Y. Ding, J. Phys. Chem. C, 2008, 112, 9673–9678 CrossRef CAS.
- R. J. Angelici, J. Organomet. Chem., 2008, 693, 847–856 CrossRef CAS.
- M. Lazar, B. L. Zhu and R. J. Angelici, J. Phys. Chem. C, 2007, 111, 4074–4076 CrossRef CAS.
- M. Lazar and R. J. Angelici, J. Am. Chem. Soc., 2006, 128, 10613–10620 CrossRef CAS.
- K. Okazaki, S. Ichikawa, Y. Maeda, M. Haruta and M. Kohyama, Appl. Catal., A, 2005, 291, 45–54 CrossRef CAS.
- M. Chen, Y. Cai, Z. Yan and D. W. Goodman, J. Am. Chem. Soc., 2006, 128, 6341–6346 CrossRef CAS.
- M. D. Hughes, Y. J. Xu, P. Jenkins, P. McMorn, P. Landon, D. I. Enache, A. F. Carley, G. A. Attard, G. J. Hutchings, F. King, E. H. Stitt, P. Johnston, K. Griffin and C. J. Kiely, Nature, 2005, 437, 1132–1135 CrossRef CAS.
- D. A. Outka and R. J. Madix, Surf. Sci., 1987, 179, 351–360 CrossRef CAS.
- B. K. Min, A. R. Alemozafar, D. Pinnaduwage, X. Deng and C. M. Friend, J. Phys. Chem. B, 2006, 110, 19833–19838 CrossRef CAS.
- R. A. Ojifinni, J. L. Gong, D. W. Flaherty, T. S. Kim and C. B. Mullins, J. Phys. Chem. C, 2009, 113, 9820–9825 CrossRef CAS.
- R. A. Ojifinni, N. S. Froemming, J. Gong, M. Pan, T. S. Kim, J. M. White, G. Henkelman and C. B. Mullins, J. Am. Chem. Soc., 2008, 130, 6801–6812 CrossRef CAS.
- J. L. Gong and C. B. Mullins, Acc. Chem. Res., 2009, 42, 1063–1073 CrossRef CAS.
- J. L. Gong, D. W. Flaherty, T. Yan and C. B. Mullins, ChemPhysChem, 2008, 9, 2461–2466 CrossRef CAS.
- B. J. Xu, X. Y. Liu, J. Haubrich and C. M. Friend, Nat. Chem., 2009, 2, 61–65.
- R. G. Quiller, X. Y. Liu and C. M. Friend, Chem.–Asian J., 2010, 5, 78–86 CrossRef CAS.
- X. Y. Liu and C. M. Friend, J. Phys. Chem. C, 2010, 114, 5141–5147 CrossRef CAS.
- B. J. Xu, X. Y. Liu, J. Haubrich, R. J. Madix and C. M. Friend, Angew. Chem., Int. Ed., 2009, 48, 4206–4209 CrossRef CAS.
- B. K. Min, X. Deng, X. Liu, C. M. Friend and A. R. Alemozafar, ChemCatChem, 2009, 1, 116 Search PubMed.
- X. Y. Liu, B. J. Xu, J. Haubrich, R. J. Madix and C. M. Friend, J. Am. Chem. Soc., 2009, 131, 5757 CrossRef CAS.
- X. Y. Liu, R. J. Madix and C. M. Friend, J. Am. Chem. Soc., 2006, 128, 14266–14267 CrossRef CAS.
- X. Y. Deng, B. K. Min, X. Y. Liu and C. M. Friend, J. Phys. Chem. B, 2006, 110, 15982–15987 CrossRef CAS.
- J. L. Gong, T. Yan and C. B. Mullins, Chem. Commun., 2009, 761–763 RSC.
- I. E. Wachs and R. J. Madix, Surf. Sci., 1978, 76, 531–558 CrossRef CAS.
- A. Andreasen, H. Lynggaard, C. Stegelmann and P. Stoltze, Appl. Catal., A, 2005, 289, 267–273 CrossRef CAS.
- M. Mavrikakis, P. Stoltze and J. K. Norskov, Catal. Lett., 2000, 64, 101–106 CrossRef CAS.
- I. N. Remediakis, N. Lopez and J. K. Norskov, Angew. Chem., Int. Ed., 2005, 44, 1824–1826 CrossRef CAS.
- R. J. Madix, Science, 1986, 233, 1159–1166 CrossRef CAS.
- X. Y. Deng and C. M. Friend, J. Am. Chem. Soc., 2005, 127, 17178–17179 CrossRef CAS.
- T. Yan, J. Gong and C. B. Mullins, J. Am. Chem. Soc., 2009, 131, 16189–16194 CrossRef CAS.
- D. A. Outka and R. J. Madix, J. Am. Chem. Soc., 1987, 109, 1708–1714 CrossRef CAS.
- B. J. Xu, L. Zhou, R. J. Madix and C. M. Friend, Angew. Chem., Int. Ed., 49, 394–398 Search PubMed.
- J. Wang, M. R. Voss, H. Busse and B. E. Koel, J. Phys. Chem. B, 1998, 102, 4693–4696 CrossRef CAS.
- J. M. Gottfried, N. Elghobashi, S. L. M. Schroeder and K. Christmann, Surf. Sci., 2003, 523, 89–102 CrossRef CAS.
- M. M. Biener, J. Biener and C. M. Friend, Surf. Sci., 2005, 590, L259–L265 CrossRef CAS.
- N. D. S. Canning, D. Outka and R. J. Madix, Surf. Sci., 1984, 141, 240–254 CrossRef CAS.
- N. Saliba, D. H. Parker and B. E. Koel, Surf. Sci., 1998, 410, 270–282 CrossRef CAS.
- G. Jones, J. G. Jakobsen, S. S. Shim, J. Kleis, M. P. Andersson, J. Rossmeisl, F. Abild-Pedersen, T. Bligaard, S. Helveg, B. Hinnemann, J. R. Rostrup-Nielsen, I. Chorkendorff, J. Sehested and J. K. Norskov, J. Catal., 2008, 259, 147–160 CrossRef CAS.
- J. K. Norskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37 CrossRef CAS.
- M. M. Biener, J. Biener and C. M. Friend, Surf. Sci., 2007, 601, 1659–1667 CrossRef CAS.
- M. M. Biener, J. Biener and C. M. Friend, Langmuir, 2005, 21, 1668–1671 CrossRef CAS.
- W. W. Gao, T. A. Baker, L. Zhou, D. S. Pinnaduwage, E. Kaxiras and C. M. Friend, J. Am. Chem. Soc., 2008, 130, 3560–3565 CrossRef CAS.
- W. W. Gao, L. Zhou, D. S. Pinnaduwage and C. M. Friend, J. Phys. Chem. C, 2007, 111, 9005–9007 CrossRef CAS.
- B. K. Min, X. Deng, D. Pinnaduwage, R. Schalek and C. M. Friend, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72 Search PubMed.
- B. K. Min, A. R. Alemozafar, M. M. Biener, J. Biener and C. M. Friend, Top. Catal., 2005, 36, 77–90 CrossRef CAS.
- B. K. Min, X. Deng, D. Pinnaduwage, R. Schalek and C. M. Friend, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 4.
- T. A. Baker, C. M. Friend and E. Kaxiras, J. Phys. Chem. C, 2009, 113, 3232–3238 CrossRef CAS.
- T. A. Baker, C. M. Friend and E. Kaxiras, J. Chem. Phys., 2009, 130, 084701 CrossRef.
- X. Liu, T. A. Baker and C. M. Friend, Dalton Trans., 2010, 39, 8521 RSC.
- T. A. Baker, B. Xu, X. Liu, C. M. Friend and E. Kaxiras, J. Phys. Chem. C, 2009, 113, 16561 CrossRef CAS.
- T. A. Baker, B. Xu, S. Jensen, C. M. Friend and E. Kaxiras, 2010, in preparation.
- T. A. Baker, C. M. Friend and E. Kaxiras, J. Chem. Theory Comput., 2010, 6, 279 CrossRef CAS.
- C. Lemire, R. Meyer, S. Shaikhutdinov and H. J. Freund, Angew. Chem., Int. Ed., 2004, 43, 118–121 CrossRef.
- M. Turner, V. B. Golovko, O. P. H. Vaughan, P. Abdulkin, A. Berenguer-Murcia, M. S. Tikhov, B. F. G. Johnson and R. M. Lambert, Nature, 2008, 454, 981–U931 CrossRef CAS.
- M. S. Chen and D. W. Goodman, Science, 2004, 306, 252–255 CrossRef CAS.
- B. Yoon, H. Hakkinen, U. Landman, A. S. Worz, J. M. Antonietti, S. Abbet, K. Judai and U. Heiz, Science, 2005, 307, 403–407 CrossRef CAS.
- M. A. Barteau and R. J. Madix, J. Am. Chem. Soc., 1983, 105, 344 CrossRef CAS.
- J. T. Roberts and R. J. Madix, J. Catal., 1993, 141, 300 CrossRef CAS.
- J. T. Roberts and R. J. Madix, Surf. Sci., 1990, 226, L71 CrossRef CAS.
- S. Hawker, C. Mukoid, J. P. Baydal and R. M. Lambert, Vacuum, 1994, 45, 275 CrossRef CAS.
- X. Lin, B. Yang, H. M. Benia, P. Myrach, M. Yulikov, A. Aumer, M. A. Brown, M. Sterrer, O. Bondarchuk, E. Kieseritzky, J. Rocker, T. Risse, H. J. Gao, N. Nilius and H. J. Freund, J. Am. Chem. Soc., 2010, 132, 7745–7749 CrossRef CAS.
- X. Lin, N. Nilius, M. Sterrer, P. Koskinen, H. Hakkinen and H. J. Freund, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 153406 CrossRef.
- M. Baron, O. Bondarchuk, D. Stacchiola, S. Shaikhutdinov and H. J. Freund, J. Phys. Chem. C, 2009, 113, 6042–6049 CrossRef CAS.
- C. C. Battaile and D. J. Srolovitz, Annu. Rev. Mater. Res., 2002, 32, 297 CrossRef CAS.
- J. D. Stiehl, T. S. Kim, S. M. McClure and C. B. Mullins, J. Phys. Chem. B, 2005, 109, 6316–6322 CrossRef CAS.
- J. A. Rodriguez, S. Ma, P. Liu, J. Hrbek, J. Evans and M. Perez, Science, 2007, 318, 1757 CrossRef CAS.
- M. Haruta, Catal. Today, 1997, 36, 153 CrossRef CAS.
- L. M. Molina and B. Hammer, Appl. Catal., A, 2005, 291, 21–31 CrossRef CAS.
- L. M. Molina, M. D. Rasmussen and B. Hammer, J. Chem. Phys., 2004, 120, 7673–7680 CrossRef CAS.
- J. C. Tully, in Modern Methods for Multidimensional Dynamics Computations in Chemistry, ed. D. L. Thompson, World Scientific, Singapore, 1998 Search PubMed.
- J. C. Tully, in Classical and Quantum Dynamics in Condensed Phase Simulations, ed. B. J. Berne, G. Ciccotti and D. F. Coker, World Scientific, Singapore, 1998 Search PubMed.
- M. R. Radeke and E. A. Carter, Annu. Rev. Phys. Chem., 1997, 48, 243 CrossRef CAS.
- D. Marx and J. Hutter, Ab Initio Molecular Dynamics: Basic Theory and Advanced Methods, Cambridge University Press, Cambridge, 2009 Search PubMed.
- R. Car and M. Parrinello, Phys. Rev. Lett., 1985, 55, 2471 CrossRef CAS.
- J. D. Stiehl, T. S. Kim, S. M. McClure and C. B. Mullins, J. Am. Chem. Soc., 2004, 126, 1606–1607 CrossRef CAS.
- K. Reuter and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 65, 035406.
- A. Valdes, Z. W. Qu, G. J. Kroes, J. Rossmeisl and J. K. Norskov, J. Phys. Chem. C, 2008, 112, 9872 CrossRef CAS.
- N. Lopez, T. V. W. Janssens, B. S. Clausen, Y. Xu, M. Mavrikakis, T. Bligaard and J. K. Norskov, J. Catal., 2004, 223, 232–235 CrossRef CAS.
- H. Falsig, B. Hvolbaek, I. S. Kristensen, T. Jiang, T. Bligaard, C. H. Christensen and J. K. Norskov, Angew. Chem., Int. Ed., 2008, 47, 4835–4839 CrossRef CAS.
- B. Hvolbaek, T. V. W. Janssens, B. S. Clausen, H. Falsig, C. H. Christensen and J. K. Norskov, Nano Today, 2007, 2, 14–18 CrossRef.
- T. V. W. Janssens, B. S. Clausen, B. Hvolbaek, H. Falsig, C. H. Christensen, T. Bligaard and J. K. Norskov, Top. Catal., 2007, 44, 15–26 CrossRef CAS.
- I. N. Remediakis, N. Lopez and J. K. Norskov, Appl. Catal., A, 2005, 291, 13–20 CrossRef CAS.
- X. Y. Liu, R. J. Madix and C. M. Friend, Chem. Soc. Rev., 2008, 37, 2243–2261 RSC.
- D. Torres, K. M. Neyman and F. Illas, Chem. Phys. Lett., 2006, 429, 86–90 CrossRef CAS.
- A. Klust and R. J. Madix, J. Am. Chem. Soc., 2006, 128, 1034 CrossRef CAS.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362–365 CrossRef CAS.
- F. W. Zemichael, A. Al-Musa, I. W. Cumming and K. Hellgardt, Solid State Ionics, 2008, 179, 1401–1404 CrossRef CAS.
- N. K. Chaki, H. Tsunoyama, Y. Negishi, H. Sakurai and T. Tsukuda, J. Phys. Chem. C, 2007, 111, 4885–4888 CrossRef CAS.
- S. Rojluechai, S. Chavadej, J. W. Schwank and V. Meeyoo, Catal. Commun., 2007, 8, 57–64 CrossRef CAS.
- M. A. Barteau and R. J. Madix, Surf. Sci., 1980, 97, 101 CrossRef CAS.
- M. Bowker, Phys. Chem. Chem. Phys., 2007, 9, 3514 RSC.
- R. A. Bennett, C. Pang, N. Perkins, R. D. Smith, P. Morrall, R. Kvon and M. Bowker, J. Chem. Phys., 2002, 106, 4688 CAS.
- M. Bowker, P. Stone, P. Morrall, R. D. Smith, R. A. Bennett, N. Perkins, R. Kvon, C. Pang, E. Fourre and M. Hall, J. Catal., 2005, 234, 172 CrossRef CAS.
- X. C. Guo and R. J. Madix, Acc. Chem. Res., 2003, 36, 471 CrossRef CAS.
- R. T. Vang, J. V. Lauritsen, E. Laegsgaard and F. Besenbacher, Chem. Soc. Rev., 2008, 37, 2191 RSC.
- K. Reuter and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 045433 CrossRef.
- C. J. Cramer, Essentials of Computational Chemistry: Theories and Models, John Wiley and Sons, 2nd edn, 2004 Search PubMed.
- J. Kohanoff, Electronic Structure Calculations for Solids and Molecules, Cambridge University Press, Cambridge, 2006 Search PubMed.
| This journal is © the Owner Societies 2011 |