Species specific isotope dilution versus internal standardization strategies for the determination of Cu, Zn-superoxide dismutase in red blood cells†‡
Y. Nuevo
Ordóñez
a,
C. L.
Deitrich
b,
M.
Montes-Bayón
a,
E.
Blanco-González
*a,
J.
Feldmann
b and
A.
Sanz-Medel
*a
aDepartment of Physical and Analytical Chemistry, University of Oviedo, C/Julián Clavería 8, 33006, Oviedo, Spain. E-mail: eblancog@uniovi.es; asm@uniovi.es
bCollege of Physical Science, Department of Chemistry, University of Aberdeen, Meston Walk, Aberdeen, UK AB24 3UE
First published on 10th December 2010
Abstract
Many proteins (more than one third) contain metal ions either within their own structures or bound to some of their active sites. These metals are involved in numerous biological processes and therefore, the quantification of metalloproteins that can serve as clinical biomarkers is of great interest. With this aim, the development of analytical strategies that permit individual (targeted) protein quantitative analysis is attempted in this work. In particular, the evaluation of different strategies for the determination of Cu, Zn-superoxide dismutase (Cu, Zn-SOD), a metalloprotein present in the first-line antioxidant defence system of the body, is conducted. The first analytical strategy is based on the use of bovine Cu, Zn-SOD as internal standard for the quantitative analysis of human Cu, Zn-SOD. For this aim, the chromatographic separation between both species (bovine and human) has been optimized according to their respective isoelectric point by anion exchange chromatography. Interestingly, the obtained results revealed a faster specific degradation of the bovine standard with respect to human SOD during sample preparation. The second strategy involves the production and evaluation of an isotopically enriched metalloprotein standard to be used as tracer in the species specific isotope dilution (SS-IDA) method by measuring the Cu associated to the protein. This is done by liquid chromatography with online inductively coupled plasma mass spectrometric (ICP-MS) detection and applied to the quantification in bovine erythrocytes. This finding is a good example to illustrate the power of SS-IDA for targeted protein quantification in respect to the commonly used alternative standards.
Introduction
A first step towards the molecular understanding of the pathogenic mechanism of oxidative stress in different diseases is to know the causing molecules of such redox imbalance or/and evaluate their consequences. The most plausible cause for oxidative stress is the presence of free radicals1 and there are some intrinsic difficulties in their direct measurement: i) the ultra-short half-life of these species (usually microseconds) due to their high reactivity and ii) many of the generated end products are in themselves reactive, although to a lesser degree. Therefore, the measurement of the effects caused by free radical action is the preferred way to evaluate oxidative stress. Among the different possible biomarkers for oxidative stress, the measurement of changes on the expression of redox controlling enzymes such as Cu, Zn-superoxide dismutase (SOD1; EC 1.15.1.1) are widely used. Additionally, point mutations in this protein and the presence of oxidative adducts are considered as specific biomarkers Alzheimer’s disease (AD),2 Parkinson’s disease (PD)3 and amyotrophic lateral sclerosis (ALS).4 Cu, Zn-SOD is a homodimeric enzyme (32 kDa) first discovered by McCord and Friedovic,5 that catalyzes the dismutation of superoxide radicals into molecular oxygen and hydrogen peroxide, which is then decomposed by catalase and glutathione peroxidase. The dismutation reaction takes place via its Cu ion redox cycle [Cu-(II)/Cu(I)], while the neighboring Zn ion plays a structural role.6Thus, Cu, Zn-SOD has been established as one of the important target biomolecules to address changes in the redox status of the organisms and to protect for oxidative stress.7 However, this can only be applied for clinical monitoring if sensitive and specific methods for Cu, Zn-SOD determination exist to quantitatively distinguish between true disease values clearly discriminated from the noise of individual biological variations. Such quantitative targeted protein analysis is generally conducted by combination of stable isotope dilution with multiple reaction monitoring (MRM) via electrospray mass spectrometry (ESI-MS).8 However, the determination of Cu, Zn-SOD in biological samples has been generally accomplished by activity assays due to the relatively high concentration of this protein reported in red blood cells (>20 mg L−1, ranging between 20 and 240 mg L−1).9,10 The levels found in serum (>100 μg L−1) require more sensitive and specific strategies such as ELISA assays that provide limits of detection low enough (about 40 ng L−1) to address small variations among individuals.11,12 In any case, the variation of the published concentration results among different methods is so extensive that no reference levels of Cu, Zn-SOD have been established yet either in serum or in red blood cells. Additionally, variations due to different pathological conditions can led to an increase of this biomarker (in, e.g. acute myelogenous leukemia or renal failure where concentration in plasma can increase from 54 ± 21 ng mL−1 in plasma to 354 ± 125 ng mL−1).13 Also, a decrease in SOD levels (in e.g., malignant lymphoma patients, chronic alcoholism or medullar tumours where the concentration decreases from 353 ± 62 ng g−1 in normal tissues to 123 ± 13 ng mL−1) can be observed.14,15
In addition to biological methods, some other alternatives have been recently proposed with the final aim of Cu, Zn-SOD absolute quantification by inductively coupled plasma mass spectrometric (ICP-MS) consisting of determining the Cu bound to the biomolecule and calculating the protein content from the known metal:protein stoichiometry. Some of these strategies are based on the appropriate gel electrophoretic separation of the protein followed by the elemental detection of the Cu and Zn on the ablated protein spots by ICP-MS.16 In order to compensate for potential losses during the analytical procedure, Feldmann et al. have used the synthesized isotopically labelled 65Cu,68Zn-SOD that permits also the absolute quantification of the protein.17,18 But the use of isotopically labelled protein standards represents a growing area of research in both molecular (labelling on the N, O or C)19 and elemental (labelling on the heteroelements such as Fe,20,21 Cu,22etc.) mass spectrometry. It is well established that the earlier in the procedure the isotopically labelled species are spiked into the sample, the higher the potential to compensate for metal losses during the whole analytical procedure, independently if the final measurement is done by ESI-MS or by ICP-MS.23
In a recent publication we have described the use of anion exchange high performance liquid chromatography (HPLC) with ICP-MS detection for Cu, Zn-SOD quantification in red blood cells using post-column isotope dilution analysis.24 This study revealed that losses of about 30% were occurring during sample preparation, specifically during the haemoglobin precipitation step. One alternative to address such losses is the use of standard additions, which is feasible in the case of Cu, Zn-SOD since the human enzyme is commercially available. However, this is a time consuming and expensive methodology since spiking at 2–3 levels of concentration have to be conducted.25 Another possibility is the use of an adequate internal standard. This possibility has been demonstrated successfully for quantification using ICP-MS and the capability of this instrument to provide almost species independent ionization of heteroelements.26 Indeed, the best results should be obtained if the internal standard were as close in structure as possible to the sought molecule and if it can be spiked at the beginning of the sample preparation.25 This is approximated in protein analysis by using bovine standards which are often available and relatively affordable.27
Thus, in this work we explored the possibility to conduct the determination of Cu, Zn-SOD in human red blood cells by using a bovine Cu, Zn-SOD as internal standard. Also the possibility of using species-specific spiking mode of isotope dilution analysis (“the perfect internal standard”) will be evaluated and both strategies critically compared.
Materials and methods
Reagents, Materials and Samples
Cu, Zn-SOD from bovine was purchased from Sigma-Aldrich (St. Louis, MO, USA). Mobile phases for HPLC containing: A) 10 mM tris-aminomethane (Merck, Darmstadt, Germany)/Acetic acid (Merck), pH 7.4 and B) A + 250 mM ammonium acetate (Merck) were prepared by dilution of the solid salts with the 18 MΩcm distilled de-ionized water (Millipore, Bedford, MA, USA). Isotopically enriched Cu and Zn solutions with relative abundances of 98.54% 65Cu and 85.99% 68Zn were prepared by dilution in water of the stock solution (604.4 μg g−1 of Cu and 1009.8 μg g−1 of Zn) obtained from Spectrascan (Teknolab A.S. Dröbak, Norway). Cut-off membrane filters (10 KDa) were obtained from Amicon-Ultra (Millipore Iberica, Madrid). The Bovine samples were provided from a farm and extracted by a veterinarian and the human samples were provided by Hospital Central of Asturias (Spain).HPLC-ICP-MS
HPLC separations were carried out using a dual piston liquid chromatographic pump (Shimadzu LC-10AD, Shimadzu Corporation, Kyoto, Japan) equipped with a sample injection valve Rheodyne, Model 7125 (Cotati, CA, USA), fitted with a 50 μL injection loop, an anion-exchange column, Mono-Q HR 5/5 (50 × 5 mm id, Pharmacia, Amersham Bioscience, Spain) and a Diode Array Detector (DAD) from Agilent Technologies (1100 Series, Waldbron, Germany). All the mobile phases were passed through a scavenger column (25 × 0.5 mm id) placed between the pump and the injection valve in order to eliminate possible metal traces present as contamination in the mobile phases. The scavenger column was packed with Kelex-100 (Schering, Germany) impregnated silica C18 material (20 μm particle size) (Bondapack, Waters Corporation, Massachusetts, USA). Specific atomic detection of Cu and Zn in the column effluent was performed using an inductively coupled plasma mass spectrometer Model 7500 from Agilent Technologies (Agilent, Tokyo, Japan) equipped with a collision cell system (ICP-(ORS)-MS).Preparation of the isotopically enriched Cu, Zn-superoxide dismutase
The isotopically enriched SOD was prepared using a combination of previously reported methods by Deitrich et al.17 and Yamazaki et al.28 In brief, the commercial protein standard (either human or bovine) was dissolved in 10 mM ammonium acetate pH 6.3. These stock solutions were then transferred into 3-mL dialysis membranes with a 5000 Da molecular weight cut-off (MWCO) (Spectra/Pro CE Flot-A-Lyzer, Spectrum Europe B.V.). Enzyme solutions were dialyzed against a 20 mM ammonium acetate buffer (pH 6.3) for 1 h at room temperature to remove low molecular weight impurities in order to obtain a purified stock solution. For preparation of the apo-enzyme, the solution was dialyzed against 20 mL of 20 mM ammonium acetate buffer (pH 3.1) containing 5 mM EDTA for 2h. Then, the dialysis buffer was changed to a 20 mM ammonium acetate buffer (pH 3.1) without EDTA (to remove the EDTA). After 2 h, the dialysis buffer was replaced with 20 mM ammonium acetate buffer (pH 6.8) and allowed to stand for 1 h, giving the apo-enzyme solution. The structure of the apo-enzyme was confirmed by MALDI-TOF (Voyager-DETM STR BiospectrometryTM Workstation, Applied Biosytems, Langen, Germany) and by ESI-MS (LC/MSD Trap System, Agilent Technologies, Waldbron, Germany).24The resulting apo-enzyme solution was then remetallated with the isotopically enriched 65Cu and 68Zn solutions. For incorporation of the isotopically enriched metals into the apo-enzyme, an excess of 68Zn and 65Cu solutions were added sequentially at different pH. First, the Zn2+ ion solution (100 μL of 1009.8 μg mL−1, pH 3.9) was added to the apo-enzyme solution and incubated at 37 °C for 4 h, thereafter mixed with the Cu2+ ion solution (250 μL of 604.4 μg mL−1, pH 8.2) and incubated at 37 °C overnight. Afterwards, the remetallated SOD1 was passed through 10 KDa cut-off filter and thoroughly washed with Milli-Q water to remove the excess of Cu and Zn. Finally the structure of the labeled species was confirmed by MALDI-TOF (intact protein analysis and trypsin digested protein). For trypsin digestion, 50 μL of the sample were treated with 50 μL urea (prepared 12 mol L−1 in 0.1 mol L−1 Tris-HCl) and 5 μL dithiotreitol (DTT, 0.2 mol L−1) to reduce the protein disulfide bonds. The mixture was centrifuged for 1 min at 700 g and was left to react for 1 h at room temperature. After this time, 20 μL of iodoacetamide (IAA, 0.2 mol L−1) were added in order to protect SH groups and the mixture was left to stand for one hour at room temperature. Finally, 20 μL more of DTT were added to consume the excess of IAA and after one hour 775 μL of Milli-Q water and 100 μL trypsin were incorporated. This cocktail was kept for 15 h at 37 °C and then the pH adjusted at about 6 with acetic acid. The peptides obtained with this procedure were then cleaned by Zip-Tip 18 and immobilized in the MALDI plate using α-ciano as matrix. The sequence coverage of the obtained peptides was 99%.
Real sample treatment
Bovine red blood cells and human red blood cells, collected with sodium citrate as anticoagulant, were hemolyzed by addition of ice cold distilled water (4.5 mL per 0.5 mL of cells) and shaking for 3 min using a vortex mixer at room temperature. After that a solution of ethanol–chloroform (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (3.75 mL per 0.5 mL of cells) was added in order to accomplish the haemoglobin precipitation. After manual shaking for 5 min, the mixture was centrifuged (12000g, 4 °C, 1 h). At last, the samples were passed through a 10 KDa cut-off filter for cleaning and preconcentration. In the case of using bovine SOD as internal standard to quantify the SOD in human red blood cells, an aliquot of the bovine Cu, Zn-SOD standard was added to the human sample of red blood cells after lysis and the sample treatment protocol was then followed. The results obtained for SS-IDA-ICP-MS were done in bovine red blood cells as proof of concept. For this purpose, an aliquot of the isotopically enriched SOD standard from bovine erythrocytes was added to the sample of bovine red blood cells after lysis and the previously described sample protocol was followed. For the calculation mass bias has been corrected by using the exponential model and dead time corrections are not necessary since the instrument performs automatic dead time correction.
:1) (3.75 mL per 0.5 mL of cells) was added in order to accomplish the haemoglobin precipitation. After manual shaking for 5 min, the mixture was centrifuged (12000g, 4 °C, 1 h). At last, the samples were passed through a 10 KDa cut-off filter for cleaning and preconcentration. In the case of using bovine SOD as internal standard to quantify the SOD in human red blood cells, an aliquot of the bovine Cu, Zn-SOD standard was added to the human sample of red blood cells after lysis and the sample treatment protocol was then followed. The results obtained for SS-IDA-ICP-MS were done in bovine red blood cells as proof of concept. For this purpose, an aliquot of the isotopically enriched SOD standard from bovine erythrocytes was added to the sample of bovine red blood cells after lysis and the previously described sample protocol was followed. For the calculation mass bias has been corrected by using the exponential model and dead time corrections are not necessary since the instrument performs automatic dead time correction.
Results and discussion
The use of bovine Cu, Zn-SOD as internal standard for human SOD1 analysis
A good internal standard should fulfil a number of prerequisites: 1) it should be as similar to the analyte in structure as possible; 2) the retention time in the chromatographic system should be as close as possible to the retention time of the analyte when an elution gradient has been used (thus, nebulization/ionization effects due to changes in the mobile phase composition can be addressed); and 3) it should be absent from the sample and chromatographically separated from interfering compounds. In this work we have evaluated the possibility of using Cu, Zn-SOD from bovine erythrocytes as an internal standard for the quantification of the SOD1 extracted from human erythrocytes. For this purpose, the first step was the evaluation of the chromatographic set-up for the separation of the two investigated standards (bovine and human standards of SOD1) under the used chromatographic conditions. Fig. 1 shows the obtained chromatogram for a mixture 1:1 of both species injected into the HPLC-ICP-MS system (the separation conditions are collected in Table 1). As can be seen, two main species can be observed in the 63Cu trace at 19 and 21 min, respectively. By injecting the individual compounds it was possible to realize that the first eluting peak (19 min) corresponds to the bovine standard while the one at 21 min corresponds to the human standard. Such good separation is due to the different isoelectric points of bovine (4.95) and human (4.75) Cu, Zn-SOD associated to the slightly different amino acid sequence. The small impurity eluting between both peaks at approximately 20 min belongs to the bovine standard and could always be detected. Once the separation between both standards was achieved, the next step was the evaluation of SOD1 from bovine erythrocytes as internal standard for quantification of human SOD1.
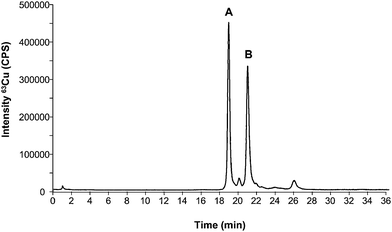 | ||
| Fig. 1 Copper profile by HPLC-ICP-MS corresponding to a mixture of a standard of bovine SOD1 (A) and human SOD1 (B) in an approximated ratio of 1:1 (400 ng ml−1 as Cu or 100 μg mL−1 as SOD). | ||
| ICP-MS Parameters | |
| Instrument | Agilent 7500 CE (ORS) |
| Rf power | 1500 W |
| Nebulizer gas flow rate | 1 L min−1 |
| m/z monitored | 63Cu, 65Cu |
| Hexapole bias | −15 V |
| QP bias | −13 V |
| HPLC Parameters | |
| Column | Mono Q HR 5/5 Anion exchange |
| Injection volume | 50 μL |
| Flow rate | 1 mL min−1 |
| Mobile phases | A) 10 mM Tris-Acetic Acid B) A + 250 mM ammonium acetate |
| Gradient | Gradient 0–100% B in 30 min (linear) |
For this purpose, two independent aliquots of the human erythrocytes lysate (5 mL) were taken and one of them was spiked with the bovine SOD1 standard (100 μL of 0.05 mg mL−1 as SOD). The obtained results are shown in Fig. 2 where it is possible to observe a good separation between species in the extracted sample without addition of internal standard (Fig. 2A, black trace) with the main peak (at 21 min) corresponding to the retention time of human erythrocytes SOD1. Fig. 2B (blue trace) shows the resulting chromatogram after spiking bovine SOD1 immediately after the lysis. In this case, the bovine standard expected to elute at 19 min can be barely detected while a new unknown Cu-containing species appears at 20 min (not present in the original sample chromatogram of Fig. 2A). In order to verify a possible shift on the retention time of the bovine standard due to the sample matrix, the same sample of Fig. 2B was spiked again with the bovine standard and re-injected in the chromatographic system. The resulting profile is shown in Fig. 2C where a good separation of the three species could be observed at 19, 20 and 21 min respectively. These initial results pointed out a possible species degradation of the bovine standard during sample preparation, since the human SOD1 present in the sample (eluting at 21 min) showed very similar intensity in all the obtained chromatograms.
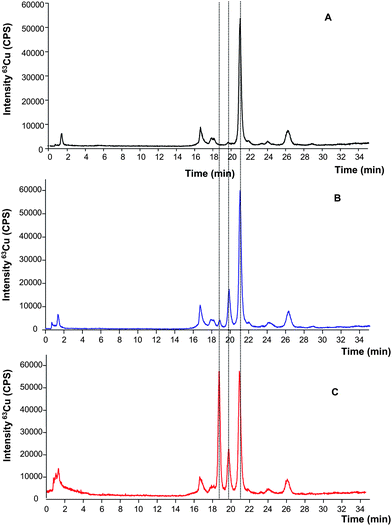 | ||
| Fig. 2 Copper signal of the SOD1 extracted from human red blood cells by HPLC-ICP-MS before spiking (A) spiked after lysis with 100 μL of 0.05 mg mL−1 bovine SOD (B) and spiked twice: after lysis and at the time of injection with bovine SOD1 at the same concentration (C). | ||
Therefore, this approach could be used for quantification only if the species degradation occurs to the same extent in the SOD from the sample and from the standard that does not seem to be this case. Since this fact has not been confirmed, these results prove the limited capability of using bovine SOD1 standard as an internal standard to quantify the SOD in human real samples. Therefore, the alternative strategy of species specific isotope dilution was evaluated further on.
Species-specific isotope dilution for human SOD1 analysis
The first part of the study was devoted to the optimization of the synthesis of the labeled species by combining previously published methodologies as described in the procedures section.17,28Fig. 3 shows the chromatographic profiles of the isotopes 63Cu and 65Cu of isotopically enriched SOD from bovine erythrocytes. The study was done first by using the bovine SOD1 standard since it can be obtained as lyophilized powder at higher concentration that the human standard. After the conditions were optimized, the remetallation protocol was also applied to the human SOD1 (data not shown). The signals of remetallated enzyme shown in Fig. 3 revealed an altered isotopic abundance when compared with those of natural standard of Cu, Zn-SOD and the isotope ratio is 63Cu/65Cu = 0.015 ± 0.002 (similar to the 65Cu standard used for the enrichment). It is important to observe that the demetallation–remetallation process has not degraded the protein since no other species can be seen in the chromatogram.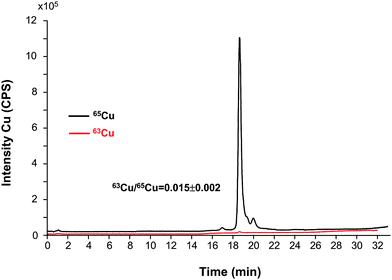 | ||
| Fig. 3 Chromatogram of the isotopically enriched bovine SOD1 obtained in the laboratory for the two copper isotopes and the corresponding isotope ratio on the SOD1 peak (250 μg mL−1 SOD obtained by dilution of the synthesized standard). | ||
The quantification of the bovine standard of isotopically enriched SOD1 was achieved by reversed post-column isotope dilution analysis using the previously developed procedure.24 For this purpose a solution containing 20 ng mL−1 of natural Cu (63Cu, 69.15%) was continuously pumped by an arm of a T piece and mixed with the eluent coming from the chromatographic column. After calculating the isotope ratio on each point of the chromatogram and applying the on-line isotope dilution equation (ESI†), it is possible to obtain the mass flow chromatogram and calculate the ng of Cu associated to the main peak in the chromatogram. This permits the indirect quantification of the SOD concentration provided that the metal/protein stoichiometry is maintained along the whole procedure (input quantities for the calculation are given in the ESI†).24,25 The concentration obtained was 0.17 ± 0.01 mg mL−1 of Cu (2.7 mM) (n = 3 injections of the diluted synthesized standard) for the isotopically enriched SOD1. Metal concentrations can be converted into protein concentrations since the stoichiometry of the protein–metal complex is known for both metals (ratio of 2:1 metal per protein). Therefore, the final concentration of isotopically enriched SOD1 is 43.5 ± 2.6 mg mL−1 (1.36 mM) (n = 3 injections). Additional experiments for spike characterization by activity measurements were not conducted since Cu has proved to be quantitatively recovered from the column and it is the catalytic center of SOD.24
In order to address if possible metal exchange between species (natural and isotopically labelled) we initially mixed the natural SOD1 standard from bovine erythrocytes (previously quantified by post column IDA) with the isotopically labelled synthesized species in an approximated 1:1 ratio. Together with metal exchanging, this experiment can be also used to corroborate the method accuracy by calculating the concentration of the natural SOD1 by species specific IDA. The concentration of Cu in the natural standard of SOD1 by this method was determined to be about 1.72 ± 0.06 mg mL−1 (27 mM or 436 mg mL−1 SOD) (n = 3 injections), a value which agrees well with the concentration obtained for the same standard by post-column IDA (1.74 ± 0.03 mg mL−1 Cu, n = 3 injections). Therefore we have proven that it should be possible to use this SOD isotopically enriched in 65Cu for the quantification of the SOD in bovine real samples since no isotopic exchange between SOD1 species can be observed. The analytical performance characteristics of the SS-IDA method are summarized in Table 2 and compared with those provided by ELISA assay. It is noteworthy that ELISA provides better sensitivity than the proposed strategy although the linear dynamic range of the SS-IDA method makes it more suitable for analysis of SOD1 in red blood cells (with concentration levels in the ppm range). In the case of serum, where the reported SOD1 concentrations are in the ng mL−1, the ELISA assay could be more suitable.
| Method | LODa/ng ml−1 | LOQa/ng mL−1 | %RSD (3 injections) | %RSD (3 lysis) | Linear range/ng mL−1a |
|---|---|---|---|---|---|
| a Expressed as concentration of SOD. | |||||
| SS-IDA HPLC-ICP-MS | 100 | 800 | 2 | 10 | 100–5000 |
| ELISA | 0.04 | 0.2 | 3 | 5 | 0.08–5 |
Once we proved that the isotopically enriched SOD could be used as a standard for species-specific IDA-ICP-MS, we applied this compound to quantify the concentration of SOD1 in erythrocytes extracted from a cow blood sample. For this aim, the isotopically labeled spike is added to the cells right after lysis and the sample processed as described in the sample treatment section. The mixture is then injected in the HPLC-ICP-MS systems. The copper chromatogram obtained is shown in Fig. 4 and as can be seen, no species degradation seems to be observed (differently to the case of using bovine SOD1 as internal standard for human SOD1). After application of the species specific isotope dilution equation (shown in the ESI†) to the peak areas in the chromatogram, the final concentration of Cu in the sample turned out to be 19.05 ± 0.03 ng mL−1 (31 mM or 4.83 μg mL−1 as SOD) (n = 3 injections) or 18 ± 1 ng mL−1 (n = 3 independent lyses). Similarly, the SOD1 from the cow red blood cells was quantified by means of post-column IDA. In this case the Cu obtained results (13.62 ± 0.02 ng mL−1 or 3.46 μg mL−1 as SOD, n = 3 injections) are lower than those obtained by species-specific IDA. Since the column recovery has proved to be quantitative for Cu and for Cu-SOD (but not for Zn) in previous studies,24 the losses (about 30%) might occur during hemoglobin precipitation and sample preconcentration steps. In any case, the obtained results corroborate the IDA principle that the earlier the spike is mixed with the sample, the more errors along the sample preparation/isolation can be compensated.
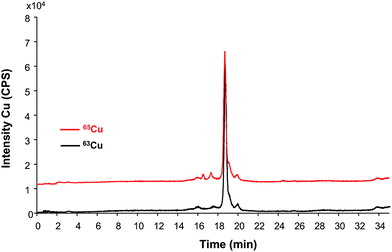 | ||
| Fig. 4 Chromatogram of the extracted SOD1 from bovine red blood cells by HPLC-ICP-MS after spiking with isotopically enriched bovine SOD1 right after lysis and used for quantitative purposes (approximately 10 μg mL−1 as SOD). Red trace is off-axes (10000 cps) for clarity. | ||
Conclusions
Two different strategies have been evaluated for quantification of Cu, Zn-SOD in red blood cells. The first one makes use of the bovine SOD species as an internal standard to quantify the human SOD1. Such a method has proved to be feasible (without observing species degradation) when the sample and the spike are mixed just before injection in the chromatographic system. However, when the spiking is conducted at the beginning of the sample treatment procedure, specific degradation of the bovine species used as standard with respect to the human SOD can be observed with time. Therefore, the next step was the evaluation of using the same species as internal standard but isotopically enriched and then species specific isotope dilution analysis. In this regard, the optimization of the isotopically enriched species synthesis permitted to obtain a fairly pure isotopically labelled standard of bovine SOD1 to be used for further quantitative studies. The SS-ID-ICP-MS allow the quantitative determination of Cu, Zn-SOD in real red blood cells of bovine samples and compensate for loses during sample preparation that turned out to be of about 30% of the total SOD1. Since lysis is a process that affects cell walls, the fact of adding the protein spike (already solubilised) before or after lysis does not affect the protein recovery from the sample. On the other hand, once the protein from the red blood cells is solubilised in the water media and the spike solution of isotopically enriched SOD is added, both species have proved to behave in the same way. Such results are in agreement with previous standard addition measurements in human SOD1 that revealed that while the chromatography used was fully quantitative the rest of the sample preparation steps were somehow prone to losses. In any case, the appropriate use of the species specific IDA method could be compared with AQUA strategies. However, a very important difference is that AQUA peptides are added into the sample after tryptic digestion of the protein to the corresponding peptides, thus, any losses of the procedure before this point can not be taken into account. When using SS-IDA, the spiking can be done right after lysis (or even before) and thus, all the losses during sample handling will have an affect in the same way on the sample protein and the spike and will therefore, be compensated.Acknowledgements
The authors want to thank the Spanish Ministry of Science for funding through the projects CTQ2007-60206/BQU and CTQ2006-02309, cofinanced through EU Feder Program. C.L.D./J.F. thanks the College of Physical Sciences and EPSRC GR/S98689/01 for financial support. The support with the instruments of Agilent Technology and Thermo-Fisher is also acknowledged.References
- K. H. Cheesman and T. F. Slater, Br. Med. Bull., 1998, 49, 481–93 Search PubMed.
- N. H. Zawia, D. K. Lahiri and F. Cardozo-Pelaez, Free Radical Biol. Med., 2009, 46, 1241–1249 CrossRef CAS.
- M. Valko, D. Leibfritz, J. Moncol, M. T. D. Cronin, M. Mazura and J. Telser, Int. J. Biochem. Cell Biol., 2007, 39, 44–84 CrossRef CAS.
- T. Sato, Y. Yamamoto, T. Nakanishi, K. Fukada, F. Sugai, Z. Zhou, T. Okuno, S. Nagano, S. Hirata, A. Shimizu and S. Sakoda, J. Neurol. Sci., 2004, 218, 79–83 CrossRef CAS.
- J. M. McCord and I. Fridovich, J. Biol. Chem., 1969, 244, 6049–6055 CAS.
- J. A. Tainer, E. D. Getzoff, K. M. Beem, J. S. Richardson and D. C. Richardson, J. Mol. Biol., 1982, 160, 181–217 CrossRef CAS.
- A. Shimizu, T. Nakanishi, M. Kishikawa and A. Miyazaki, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2002, 776, 15–30 CrossRef CAS.
- P. Picotti, O. Rinner, R. Stallmach, F. Dautel, T. Farrah, B. Domon, H. Wenschuh and R. Aebersold, Nat. Methods, 2010, 7, 43–46 CrossRef CAS.
- A. Concetti, P. Massei, G. Rotilio, M. Brunori and E. A. Rachmileqitz, J. Lab. Clin. Med., 1976, 87, 1057–1064 CAS.
- M. Minami and M. Yoshikawa, Clin. Chim. Acta, 1979, 92, 337–342 CrossRef CAS.
- T. Adachia, M. Nakamuraa, H. Yamadab, A. Futenmab, K. Katob and K. Hirano, Clin. Chim. Acta, 1994, 229, 123 CrossRef.
- P. Mondola, M. Bifulco, R. Serù, T. Annella, M. R. Ciriolo and M. Santillo, FEBS Lett., 2000, 467, 57–60 CrossRef CAS.
- K. Pawlaka, D. Pawlakb and M. Mysliwieca, Clin. Biochem., 2005, 38, 700–705 CrossRef.
- Y. Sun, L. W. Oberley and Y. Li, Clin. Chem., 1988, 34, 497–500 CAS.
- K. Iwase, A. Nagasaka, K. Kato, A. Itoh, S. Jimbo, Y. Hibi, N. Kobayashi, H. Yamamoto, T. Seko and K. Miura, J. Surg. Res., 2006, 135, 150–155 CrossRef CAS.
- L. M. Wang, J. S. Becker, Q. Wu, M. F. Oliveira, F. A. Bozza, A. L. Schwager, J. M. Hoffman and K. A. Morton, Metallomics, 2010, 2, 348–353 RSC.
- C. L. Dietrich, A. Raab, B. Pioselli, J. Thomas-Oates and J. Feldmann, Anal. Chem., 2007, 79, 8381–8390 CrossRef CAS.
- C. L. Deitrich, S. Braukmann, A. Raab, C. Munro, B. Pioselli, E. M. Krupp, J. E. Thomas-Oates and J. Feldmann, Anal. Bioanal. Chem., 2010, 397, 3515 CrossRef CAS.
- T. Geiger, J. Cox, P. Ostasiewicz, J. R. Wisniewski and M. Mann, Nat. Methods, 2010, 7, 383–385 CrossRef CAS.
- M. E. Del Castillo, M. Montes-Bayón and A. Sanz-Medel, Anal. Chem., 2006, 82, 8218–8226 CrossRef.
- M. Hoppler, C. Zeder and T. Walczyk, Anal. Chem., 2009, 81, 7368–7372 CrossRef CAS.
- C. F. Harrington, D. S. Vidler, M. J. Watts and J. F. Hall, Anal. Chem., 2005, 77, 4034–4041 CrossRef CAS.
- J. Meija and Z. Mester, Anal. Chim. Acta, 2008, 607, 115–125 CAS.
- Y. Nuevo-Ordóñez, M. Montes-Bayón, E. González-Blanco and A. Sanz-Medel, Anal. Chem., 2010, 82, 2387–2394 CrossRef.
- A. Sanz-Medel, M. Montes-Bayón, M. R. Fernández de la Campa, J. Ruiz Encinar and J. Bettmer, Anal. Bioanal. Chem., 2008, 390, 3–16 CrossRef CAS.
- A. Pereira, J. Ruiz Encinar, M. Carrascal, J. Abian and A. Sanz-Medel, Anal. Chem., 2008, 80, 1777–1787 CrossRef.
- H. Yang, Y. Y. Zhang and U. Poschl, Anal. Bioanal. Chem., 2010, 397, 879–886 CrossRef CAS.
- Y. Yamazaki and T. Takao, Anal. Chem., 2008, 80, 8246–8252 CrossRef CAS.
Footnotes |
| † This article is part of a themed issue highlighting outstanding and emerging work in the area of speciation. |
| ‡ Electronic supplementary information (ESI) available: Supplementary information. See DOI: 10.1039/c0ja00075b |
| This journal is © The Royal Society of Chemistry 2011 |