Arsenic preconcentration viasolid phase extraction and speciation by HPLC-gradient hydride generation atomic absorption spectrometry†‡
Yong
Tian
a,
Ming-Li
Chen
a,
Xu-Wei
Chen
a,
Jian-Hua
Wang
*a,
Yoshihiro
Hirano
b,
Hideyuki
Sakamoto
b and
Toshihiro
Shirasaki
b
aResearch Center for Analytical Sciences, Northeastern University, Box 332, Shenyang, 110819, China. E-mail: jianhuajrz@mail.neu.edu.cn; Fax: +86 24 83676698; Tel: +86 24 83688944
bHitachi High-Technologies Corporation, Hitachinaka, Ibaraki 312-8504, Japan
First published on 16th November 2010
Abstract
A novel method for arsenic speciation is developed by interfacing solid phase preconcentration-liquid chromatography (LC) separation-gradient hydride generation (GHG)-quartz flame atomic absorption spectrometry (QFAAS). A MnO2 mini-column is used to preconcentrate the arsenic species of As(III), As(V), MMA and DMA, during which process, As(III) is converted to As(V)viaoxidation by MnO2, while other species remain unchanged. The recovery of As(V) (i.e., the total amount of arsenate and arsenite in the original sample), MMA and DMA from the MnO2 mini-column is facilitated by tetramethylammonium hydroxide (TMAH). After LC separation with C30 columns, arsenic species in the eluate are subject to gradient hydride generation with detection by QFAAS. On the other hand, cellulose fibre selectively adsorbs the chelating complex between As(III) and ammonium pyrrolidine dithiocarbamate (APDC). After elution with HNO3, As(III) in the original sample is quantified by graphite furnace atomic absorption spectrometry (GFAAS), and the amount of As(V) is obtained by subtraction. A sample volume of 2.0 mL derives enrichment factors of 14.0–19.2 for the arsenic species. By injecting 20 μL of eluate into the LC system (the eluate of As(III)-PDC complex is injected into the GFAAS), detection limits of 0.019, 0.33, 0.39, 0.62 μg L−1 are obtained for As(III), As(V), MMA and DMA respectively. RSDs of less than 4.2% are achieved at the level of 2 μg L−1 for As(V), MMA, DMA and 1 μg L−1 for As(III). The procedure is evaluated by speciating arsenic in snow water and Hijiki samples.
Introduction
Nowadays, arsenic has been highly concerned as a toxin and carcinogen.1–3 It is known that the various arsenic species exhibit very different toxic effects on human health, e.g., the inorganic arsenic species are much more toxic than their organic counterparts.4,5 In this respect, it is no longer sufficient to evaluate the toxic effects or environmental impacts of arsenic by determining its total amount in various samples. Consequently, the differentiation of arsenic species or their speciation has been attracting more and more attentions since the last decade. The previous reports or observations have illustrated large discrepancies among the contents of the arsenic species, e.g., arsenite (As(III)), arsenate (As(V)), monomethylarsonic acid (MMA) and dimethylarsinic acid (DMA) in real samples. The concentrations of some species are quite high while those of the others are very low.6–8 The accurate quantification of ultra-trace amounts of arsenic species from real sample matrixes has been very much challenging, even with some of the highly sensitive detectors, e.g., inductively coupled plasma mass spectrometry (ICP-MS).9–11 From the practical point of view, the presence of complex concomitant sample matrixes tends to seriously deteriorate the detection capability for ultra-trace arsenic species. At the same time, the sample matrixes also degrade significantly the performance and the life-time of the HPLC column.7,11 Considering that there is a large discrepancy among the concentrations of arsenic species in real samples, it is generally necessary to inject a larger sample volume into the HPLC system in order to achieve reasonable sensitivities for the arsenic species of interest.12–15 This further exaggerates the effect of sample matrixes. We have recently proposed a gradient hydride generation protocol to improve the hydride generation efficiency of arsenic species and thus their detection sensitivities.16 However, this approach still suffers deficient sensitivity when handling ultra-trace amounts of arsenic species. From this point of view, the employment of an appropriate sample pretreatment procedure, e.g., separation and preconcentration, prior to HPLC separation is in high demand, in order to improve the sensitivity for arsenic detection and in the meantime to eliminate the adverse effect of the sample matrixes.17 A literature search reveals that there are quite a few sample pretreatment procedures focused on inorganic arsenic species,18,19 however, investigations dedicated to the simultaneous separation and preconcentration of inorganic and organic arsenic species have been scarcely reported.20Metal hydroxides and metal oxides have been employed for the adsorption and removal of arsenic.21,22 Among this category of materials, manganese dioxide (MnO2) has been identified as an arsenic demineralizer in sewage cleaning,23 and it exhibits favorable adsorption capacity for arsenic species. However, arsenic species tend to strongly retain on the surface of the MnO2, thus desorption/recovery of the arsenic species is very difficult even in high concentrations of strong acidic or basic medium with a limited volume of ca. 100–200 μL. Therefore, it is critical to find a suitable stripping reagent or eluent for the effective recovery of the adsorbed arsenic species on the MnO2 surface. At the same time, the eluent should be preferentially compatible to the HPLC system, i.e., it is better to be one of the components of the mobile phase during the HPLC separation process.
In the present work, we successively demonstrated that the arsenic species preconcentrated onto the MnO2 surface, e.g., As(III), As(V), MMA and DMA, could be readily recovered by tetramethylammonium hydroxide (TMAH) which is a component of the mobile phase for HPLC separation of the arsenic species. By coupling the preconcentration unit with HPLC separation and gradient hydride generation followed by quartz flame atomic absorption spectrometric detection, the sensitivities for the speciation of the four arsenic species were significantly improved.
Experimental
Instrumentation
A FIAlab-3000 sequential injection system equipped with two syringe pumps (with volumes of 5 mL and 2.5 mL respectively), an 8-port selection valve (FIAlab Instruments, Bellevue, WA, USA) is employed for fluidic delivery and preconcentration of the arsenic species of interest. All the external channels are made of PTFE tubing (0.8 mm i.d.) connected to the multi-port selection valve with PEEK nuts/ferrules (Upchurch Scientific). A mini-column packed with cellulose fibre is used for the adsorption and preconcentration of As(III)-ammonium pyrrolidine dithiocarbamate (APDC) complex. Another mini-column of MnO2 particles is used for the preconcentration of As(III), As(V), monomethylarsonic acid (MMA) and dimethylarsinic acid (DMA). A homemade online filter with 0.45 μm mixed cellulose ester membrane is used for the filtration of the fluids before injecting into the HPLC system.A L-2130 pump and a L-2300 column oven (Hitachi High-Technologies Corporation, Japan) with Develosil RPAQUEOUS-AR-5 columns (Φ6 × 250 mm and Φ6 × 150 mm) (Nomura Chemical Co., Ltd., Japan) are employed for chromatographic separation of the arsenic species. An injection valve (Rheodyne 7725i, Rheodyne, Cotati, CA, USA) with a 20 μL sample loop made of PEEK tubing (0.5 mm i.d.) is employed for the introduction of sample solution. A HITACHI Z-2000 atomic absorption spectrophotometer (Hitachi High-Technologies Corporation, Japan) integrating a graphite furnace atomizer and a flame atomizer is used for the detection of arsenic. The flame also facilitates the operation of a quartz atomizer.
A Minipuls evolution peristaltic pump (Gilson, France) is used for the delivery of hydrochloric acid and NaBH4 solutions to facilitate the hydride generation process. 20 μL of the LC effluent is directed into the reaction system via a piece of PEEK tubing (0.13 mm i.d.). A water bath controlled at 60 °C is used to speed up the hydride generation reaction and improve the hydride generation efficiency of the arsenic species, followed by a cooling water bath to regulate the temperature of the reaction mixture before entering the gas–liquid separator. The hydrides after separation are swept by an argon stream to flow into the T-type quartz atomizer for arsenic quantification.
A Thermo Model 868 pH meter is used to measure pH values. A single computer is used to control the sequential injection sample processing system, the HPLC system and the atomic absorption spectrometer.
Fig. 1 illustrates the flow manifold of the entire system. The left is the sample pretreatment and preconcentration unit, while the right part denotes the HPLC separation and gradient hydride generation system interfacing to quartz flame atomic absorption spectrometry thorough a gas–liquid separator.
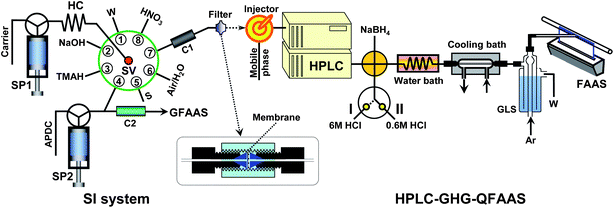 | ||
| Fig. 1 Flow manifold for the arsenic speciation system integrating preconcentration-LC separation-gradient hydride generation-quartz flame atomic absorption spectrometry. SP1, SP2: syringe pumps; HC: holding coil; C1: MnO2 mini-column; C2: cellulose fibre mini-column; SV: selection valve; W: waste; S: sample; GLS: gas liquid separator. Valve position I: 6 mol L−1HCl for the hydride generation of As(V), As(III) and MMA. Valve position II: 0.6 mol L−1HCl for the hydride generation of DMA. | ||
Materials and reagents
All chemicals used are at least of analytical reagent grade, which are received from Sinopharm Chemical Reagent Co. (Shanghai, China) or otherwise specified. De-ionized water of 18.2 MΩ cm is used throughout.Arsenic stock solutions of 1000 mg L−1 are prepared by dissolving appropriate amounts of sodium arsenite, disodium hydrogen arsenate heptahydrate, monomethylarsonic acid (MMA, Wako Pure Chemical Ind., Ltd, Japan) and dimethylarsinic acid (DMA, Wako Pure Chemical Ind., Ltd, Japan) in de-ionized water.
The reagents used in the mobile phase of LC separation process in the present study include sodium 1-butanesulfonate (Sigma-Aldrich, Switzerland), malonic acid, tetramethylammonium hydroxide, ammonium tartrate and methanol. Other reagents employed are sodium tetrahydroborate, hydrochloric acid and L-cysteine, ammonium pyrrolidine dithiocarbamate, nitric acid, magnesium nitrate hexahydrate and sodium hydroxide. In practice, NaBH4 solutions of desired concentrations in 0.5% (m/v) NaOH were prepared daily. De-ionized water is used as a carrier solution throughout the experiments.
Manganese dioxide (Sinopharm Chemical Reagent Co., Shanghai, China) is used for the preconcentration of As(III), As(V), MMA and DMA, while wood cellulose fibre (Lvjian Industry and Trade Co., Yixing, China) is used for the adsorption of As(III)-PDC complex.
Sample pretreatment
Mini-column packing
On-line preconcentration of arsenic species
400 μL of air and 100 μL of TMAH (0.04M) are sequentially aspirated into the holding coil at 100 μL s−1. The TMAH zone is thereafter directed to flow through C1 at 15 μL s−1via port 7 to collect the arsenic species adsorbed on the surface of the MnO2 mini-column, and it is further pushed to flow through the filter. The filtrate is collected and 20 μL of which is injected into the LC system to facilitate the ensuing separation-gradient hydride generation process, and the arsenic species are finally detected by quartz flame atomic absorption spectrometry. At the end of the preconcentration process, the carrier left in the syringe pump SP1 is directed to waste via port 1. The LC-GHG-QFAAS parameters during the entire operation process are illustrated in Table S1, ESI.†
Refreshing of the mini-columns
Whenever necessary, the MnO2 surface is refreshed by passing 800 μL of NaOH (2.0 mol L−1) and 700 μL of HNO3 (1.0 mol L−1) solutions through the mini-column, followed by rinsing with sufficient amounts of de-ionized water (ca. 1500 μL) until neutral effluent is obtained, and it is finally evacuated with air. As for the cellulose fibre mini-column, the refreshing is done by passing 200 μL of HNO3 (1.0 mol L−1) through it, followed by rinsing with sufficient amount of de-ionized water and evacuation with air. After 100 times operation of adsorption/elution of the arsenic species by following the above mentioned procedure, no obvious deterioration of the performance of the MnO2 mini-column has been observed.Results and discussion
Preconcentration of the arsenic species on MnO2 mini-column
In the present work, MnO2 has been demonstrated to be a suitable adsorbent for retaining the arsenic species, e.g., As(III), As(V), MMA and DMA. As these species are strongly bound onto the surface of MnO2, we have failed to achieve a reasonable recovery or de-sorption in strong acidic or alkali medium, e.g., HNO3 and NaOH solutions of various concentrations. This observation is in accordance with a previous report.24 Generally, considering that the eluate is used for the ensuing LC separation, and thus an ideal stripping reagent or eluent should be one of the components involved in the mobile phase of the LC separation system, in order to minimize the degradation of the HPLC column by the extra reagent introduced. In the present case, we are lucky enough to find that tetramethylammonium hydroxide (TMAH), a component of the mobile phase, entails favorable recovery of the retained arsenic species on the surface of the MnO2 mini-column. The results indicated that 100 μL of a 0.04 mol L−1TMAH solution provides elution efficiencies of 89%, 89%, 87%, 70% for As(III), As(V), MMA and DMA. This observation might be attributed partly to the solubilization effect of TMAH for metal ions,25 which is a driving force for the dissolution of arsenic species from the MnO2 surface.The experiments also indicated that the amount of MnO2 used for packing the mini-column is a critical factor for governing the column performance. Fig. 2 illustrates a strong dependence of the adsorption efficiencies of arsenic species on the quantity of MnO2 used for the mini-column packing. A quantity of less than 120 mg of MnO2 results in an incomplete adsorption of the arsenic species, e.g., an adsorption efficiency of less than 91% for DMA is obtained, and those for the other species are between 96%–100%. The increase of the MnO2 amount for the mini-column packing improves the retention of the arsenic species, e.g., 140 mg of MnO2 entails an adsorption efficiency of ca. 99% for DMA. On the other hand, when a larger amount of MnO2 is used, e.g., more than 160 mg, it tends to creates flow resistance in the mini-column and thus causes deterioration for the adsorption of the arsenic species, and a leakage of the syringe pump is observed at a higher flow rate of 30 μL s−1. In addition, too much absorbent causes a further challenge to the elution step, which results in the decline of the elution efficiency, or more eluent is required in order to maintain a reasonable recovery but in the meantime the arsenic species in the effluent is diluted and thus deteriorates the sensitivity for the arsenic species. For further investigations, 140–160 mg of MnO2 is used for packing the mini-column.
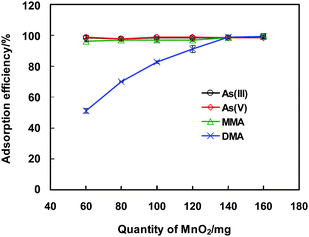 | ||
| Fig. 2 The dependence of adsorption efficiency of arsenic species on the quantity of MnO2 for mini-column packing. 20 μg L−1 of As(III), As(V), MMA and DMA are used. The conditions for preconcentration and elution are given in Table 1, and those for LC-GHG-QFAAS and GFAAS are illustrated in Table S1 and Table S2 (ESI† respectively. | ||
The acidity of the sample solution plays an important role in the preconcentration of arsenic species on the MnO2 mini-column, as illustrated in Fig. 3. It is obvious that within a reasonable range of pH 1–4, virtually complete adsorption of the four arsenic species is achieved, except for that of DMA which gives rise to an adsorption efficiency of ca. 90% at pH 1. When further increasing the pH value to exceed 4 for As(V) and DMA, 5 for As(III) and MMA, a significant drop of the adsorption efficiency of the arsenic species is observed. For further investigations, the acidity of the sample solution is fixed at pH 3.
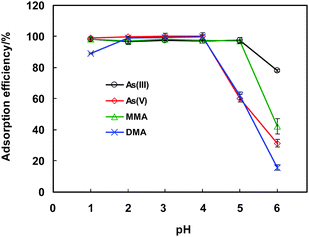 | ||
| Fig. 3 pH dependence of the adsorption efficiencies of the four arsenic species. 20 μg L−1 of As(III), As(V), MMA and DMA are employed. The conditions for preconcentration and elution are given in Table 1, and those for LC-GHG-QFAAS and GFAAS are illustrated in Table S1 and Table S2 (ESI†), respectively. | ||
Other experimental parameters including the concentration, volume and flow rate of the sample and various reagents during the adsorption, stripping/elution and mini-column refreshing processes are summarized in Table 1.
Conversion of As(III) to As(V) on the MnO2 mini-column
After the preconcentration process under the aforementioned experimental conditions, 20 μL of the collected effluent containing the arsenic species is injected into the LC system for chromatographic separation, followed by gradient hydride generation and quartz flame atomic absorption spectrometric detection. Before entering the LC system, the effluent is filtered through an on-line filter with a 0.45 μm mixed cellulose ester membrane, as illustrated in Fig. 1. The gradient hydride generation is performed as described in the previous work,16 that is a 6 mol L−1HCl is used for the hydride generation of As(V) and MMA, when the signal from MMA has been recorded, a stream of 0.6 mol L−1HCl as hydride generation medium is introduced for DMA. 2% (m/v) of L-cysteine is included in the HCl solution.16 During the gradient hydride generation process, a fixed concentration of NaBH4 at 1.0% (m/v) is used for all the arsenic species. The recorded chromatograms are illustrated in Fig. 4. It is obvious that baseline separation of As(III), As(V), MMA and DMA is obtained for the standard solution containing 20 μg L−1 of each arsenic species. After preconcentration the chemical oxidation states for As(V), MMA and DMA remain unchanged. In the meantime, a significant enhancement on the signal is achieved, which indicates that MnO2 is a suitable medium for the preconcentration of these three arsenic species. On the other hand, however, the complete disappearance of the peak signal attributed to As(III) in the effluent well denotes the conversion of As(III) to As(V), with clear demonstration by the obvious increase of the signal of As(V) as compared to those by MMA and DMA. This observation is well agreed with the previous reports.26,27 In addition, when further compared with the chromatographs in Fig. 5 recorded in the absence of As(III), it can be seen that the absorbance signal for As(V) in Fig. 4 is indeed ca. twice as high as that obtained for same amount of As(V), i.e., 20 μg L−1, in Fig. 5. This indicates that the conversion of As(III) into As(V) on the surface of MnO2 is virtually complete. This is further demonstrated by Fig. 6 showing a complete overlap of the calibration graphs for As(III) and As(V) after their preconcentration on the MnO2 mini-column followed by LC separation and quartz flame atomic absorption spectrometric detection. It shows that there is no significant difference between the two calibration graphs, or complete conversion of As(III) into As(V) can be concluded. In another words, the results imply that same amount of As(III) and As(V) give rise to equal absorbance signal after preconcentration by the MnO2 mini-column.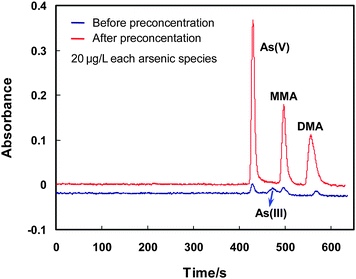 | ||
| Fig. 4 The chromatograms achieved for As(III), As(V), MMA and DMA before and after preconcentration by the MnO2 mini-column. 20 μg L−1 of As(III), As(V), MMA and DMA are employed. The conditions for preconcentration and elution are given in Table 1, and those for LC-GHG-QFAAS are illustrated in Table S1, ESI.† | ||
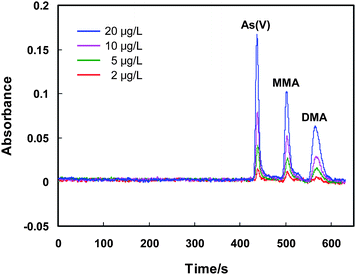 | ||
| Fig. 5 The chromatograms for various concentrations of As(V), MMA and DMA after preconcentration on the MnO2 mini-column. The conditions for preconcentration and elution are given in Table 1, and those for LC-GHG-QFAAS are illustrated in Table S1, ESI.† | ||
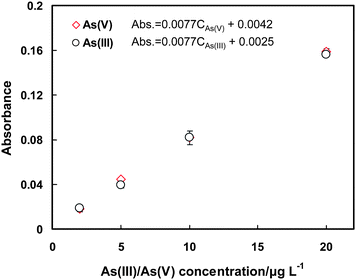 | ||
| Fig. 6 The calibration graphs for As(III) and As(V) after preconcentration on the MnO2 mini-column. Test of significance shows no significant difference between the absorbance for same amount of As(III) and As(V). Uncertainties represent standard deviations (n = 2). The conditions for preconcentration and elution are given in Table 1, and those for LC-GHG-QFAAS are illustrated in Table S1, ESI.† | ||
At this stage, the total inorganic arsenic, MMA and DMA can be differentiated with MnO2 preconcentration followed by LC separation-gradient hydride generation-quartz flame atomic absorption spectrometric detection. However, in order to differentiate the two inorganic arsenic species, an additional approach should be included to perform selective separation of either As(III) or As(V) and facilitate their ensuing speciation.
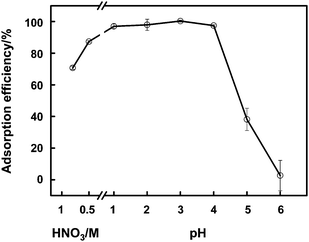 | ||
| Fig. 7 pH dependence of the adsorption efficiency of As(III)-PDC complex onto a cellulose fibre mini-column. 20 μg L−1As(III) is used. The conditions for preconcentration and elution are given in Table 2, and those for GFAAS detection are illustrated in Table S2, ESI.† | ||
In addition, we have scrutinized the dependence of the adsorption, stripping/elution of the As(III)-PDC complex on the various experimental parameters including the concentration, volume as well as flow rate of the sample solution and the reagents used. These are summarized in Table 2. After adsorption, the recovery of the As(III)-PDC complex retained on the cellulose fibre mini-column is preferentially done by using 1 mol L−1nitric acid, providing an elution efficiency of 96%.
 | ||
| Fig. 8 The preconcentration of As(III), As(V), MMA and DMA on a cellulose fibre mini-column. The retained arsenic species are recovered by using 1.0 mol L−1HNO3 and quantified with GFAAS. The conditions for preconcentration and elution are given in Table 2, and those for GFAAS detection are illustrated in Table S2, ESI.† | ||
Analytical performance and practical applications of the present system
After preconcentration, we inject 20 μL of the collected effluent into the LC system for chromatographic separation followed by gradient hydride generation-quartz flame atomic absorption spectrometric detection. The characteristic performance data of the present analytical system are summarized in Table 3. Enrichment factors of 17.0, 16.7, 14.0, 19.2 are achieved for As(V), MMA, DMA and As(III), giving rise to detection limits of 0.33, 0.39, 0.62, 0.019 μg L−1 for As(V), MMA, DMA and As(III) respectively. As a comparison to the previous report by using gradient hydride generation without preconcentration,16 the present system offers improved sensitivities for the detection of the four arsenic species, even at a much smaller consumption of the effluent for LC separation, i.e., 20 μL versus 100 μL in the previous work, which is preferential for the improvement on the lifetime of the LC column. The overall sensitivities for the arsenic species can be further improved by using a larger volume of effluent during the LC separation process.| As(V) a | MMAa | DMAa | As(III) b | |
|---|---|---|---|---|
| a preconcentration by the MnO2 mini-column followed by LC-GHG-QFAAS. b preconcentration by the cellulose fibre mini-column with detection by GFAAS. c LOD = 3HNC/H. HN: peak height of the baseline noise; C: concentration of an arsenic species; H: peak height of an arsenic species. d LOD = 3σ/K (n = 10). e RSD (%, 1 μg L−1, n = 8). | ||||
| Injection volume/μL | 20 | |||
| Linear range/μg L−1 | 1–40 | 1–40 | 2–40 | 0.1–2.0 |
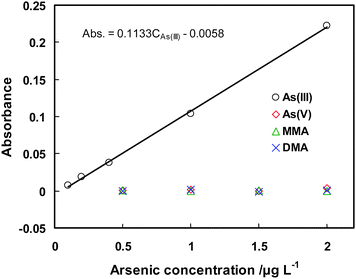
| Regression equations | A = 0.0114CAs(V) + 0.0034 | A = 0.0098CMMA + 0.0027 | A = 0.0051CDMA + 0.0034 | A = 0.1133CAs(III) -0.0058 |
| Enrichment factors | 17.0 | 16.7 | 14.0 | 19.2 |
| LODs/μg L−1 | 0.33c | 0.39c | 0.62c | 0.019d |
| RSD (%, 2 μg L−1, n = 8) | 4.2 | 2.9 | 3.9 | 2.7e |
| Sampling frequency/h−1 | 5 | 5 | 5 | 7 |
| LODs for GHG without preconcentration (ref. 16) | 0.9 | 1.4 | 1.6 | 1.4 |
We have applied this system for the speciation of arsenic in snow water and Hijiki samples, and the results are given in Table 4 and Table 5. It is obvious that only As(V) is found in the snow water sample. The analysis results for the two Hijiki samples are compared with those obtained previously by using gradient hydride generation without preconcentration.16 We can see that reasonable agreements are achieved between the two methods for the speciation of As(V), MMA and DMA, although MMA is not found in either methods. Previously, the sensitivity for As(III) by gradient hydride generation is not high enough for its detection in real samples, and thus As(III) is not found in the Hijiki samples in ref. 16. The present procedure provides a much improved sensitivity for As(III), thus a certain amount of As(III) has been identified from both Hijiki samples. It is obvious that the concentrations of As(III) in the Hijiki samples are indeed lower than the detection limit as reported in ref. 16.
| Species | Found/μg L−1 | Spiked/μg L−1 | Found/μg L−1 | Rec. (%) |
|---|---|---|---|---|
| a preconcentration by the MnO2 mini-column followed by LC-GHG-QFAAS. b preconcentration by the cellulose fibre mini-column with detection by GFAAS. Mean value ± 95% confidence interval (n = 3). c n.d: not detected. | ||||
| As(V) a | 0.5 ± 0.4 | 5.0 | 5.3 ± 0.5 | 96 |
| MMAa | n.d.c | 5.0 | 5.2 ± 0.6 | 104 |
| DMAa | n.d. | 5.0 | 5.4 ± 0.2 | 108 |
| As(III) b | n.d. | 1.0 | 1.06 ± 0.02 | 106 |
| Species | Hijiki 1 | Hijiki 2 | ||
|---|---|---|---|---|
| Found by this method/μg g−1 | Found in ref. 16/μg g−1 | Found by this method/μg g−1 | Found in ref. 16/μg g−1 | |
| a preconcentration by the MnO2 mini-column followed by LC-GHG-QFAAS. b preconcentration by the cellulose fibre mini-column with detection by GFAAS. Mean value ± 95% confidence interval (n = 3). c n.d: not detected. | ||||
| As(V) a | 50.4 ± 4.8 | 56.4 ± 5.8 | 53.6 ± 9.2 | 60.8 ± 13.2 |
| MMAa | n.d.c | n.d. | n.d. | n.d. |
| DMAa | 2.8 ± 0.9 | 2.6 ± 0.2 | 2.7 ± 0.4 | 2.4 ± 0.5 |
| As(III) b | 0.33 ± 0.03 | 0.42 ± 0.06 | n.d. | n.d. |
For the snow water sample, satisfactory spiking recoveries are achieved for the arsenic species of interest.
Conclusions
The use of a MnO2 mini-column hyphenating with sequential injection system facilitates preconcentration of arsenite, arsenate, monomethylarsonic acid (MMA) and dimethylarsinic acid (DMA). An additional cellulose fibre mini-column selectively adsorbs the arsenite-ammonium pyrrolidine dithiocarbamate (APDC) complex in the presence of the other arsenic species. By coupling preconcentration with chromatographic separation-gradient hydride generation-quartz flame atomic absorption spectrometry further improves the sensitivity for arsenic speciation as compared to the previously reported procedure based on merely gradient hydride generation. In addition, the use of a much smaller effluent volume, i.e., 20 μL, for LC separation improves the lifetime of the LC column.Acknowledgements
This work is supported by the Natural Science Foundation of China (20805004, key project 20635010, National Science Fund for Distinguished Young Scholars 20725517 and Major International Joint Research Project 20821120292) and the Fundamental Research Funds for the Central Universities (N090405004, N090105001). We also highly appreciate the technical assistance and financial support from Hitachi High-Technologies.References
- D. J. Thomas, M. Styblo and S. Lin, Toxicol. Appl. Pharmacol., 2001, 176, 127–144 CrossRef CAS.
- K. Straif, L. Benbrahim-Tallaa, R. Baan, Y. Grosse, B. Secretan, F. El Ghissassi, V. Bouvard, N. Guha, C. Freeman, L. Galichet, V. Cogliano and W. H. O. I. A. R. C. M. Workin, Lancet Oncol., 2009, 10, 453–454 CrossRef.
- Y. Chen, F. Parvez, M. Gamble, T. Islam, A. Ahmed, M. Argos, J. H. Graziano and H. Ahsan, Toxicol. Appl. Pharmacol., 2009, 239, 184–192 CrossRef CAS.
- X. J. Wang, Z. Sun, W. Chen, K. E. Eblin, J. A. Gandolfi and D. D. Zhang, Toxicol. Appl. Pharmacol., 2007, 225, 206–213 CrossRef CAS.
- M. Styblo, L. M. Del Razo, L. Vega, D. R. Germolec, E. L. LeCluyse, G. A. Hamilton, W. Reed, C. Wang, W. R. Cullen and D. J. Thomas, Arch. Toxicol., 2000, 74, 289–299 CrossRef CAS.
- Y. J. Zavala, R. Gerads, H. Gürleyük and J. M. Duxbury, Environ. Sci. Technol., 2008, 42, 3861–3866 CrossRef CAS.
- V. Nischwitz and S. A. Pergantis, Anal. Chem., 2005, 77, 5551–5563 CrossRef CAS.
- P. N. Williams, A. H. Price, A. Raab, S. A. Hossain, J. Feldmann and A. A. Meharg, Environ. Sci. Technol., 2005, 39, 5531–5540 CrossRef CAS.
- T. Nakazato and H. Tao, Anal. Chem., 2006, 78, 1665–1672 CrossRef CAS.
- S. K. V. Yathavakilla, M. Fricke, P. A. Creed, D. T. Heitkemper, N. V. Shockey, C. Schwegel, J. A. Caruso and J. T. Creed, Anal. Chem., 2008, 80, 775–782 CrossRef CAS.
- J. Lintschinger, P. Schramel, A. Hatalak-Rauscher, I. Wendler and B. Michalke, Fresenius J. Anal. Chem., 1998, 362, 313–318 CrossRef CAS.
- F. A. Duarte, J. S. F. Pereira, M. F. Mesko, F. Goldschmidt, É. M. D. Flores and V. L. Dressier, Spectrochim. Acta, Part B, 2007, 62, 978–984 CrossRef.
- P. Niedzielski, Anal. Chim. Acta, 2005, 551, 199–206 CrossRef CAS.
- J. L. Gómez-Ariza, D. Sánchez-Rodas, I. Giráldez and E. Morales, Talanta, 2000, 51, 257–268 CrossRef CAS.
- R. T. Gettar, R. N. Garavaglia, E. A. Gautier and D. A. Batistoni, J. Chromatogr., A, 2000, 884, 211–221 CrossRef CAS.
- Y. Tian, M. L. Chen, X. W. Chen, J. H. Wang, Y. Hirano, H. Sakamoto and I. Setsu, J. Anal. At. Spectrom., 2010, 25, 48–54 RSC.
- K. Wrobel, K. Wrobel and J. A. Caruso, Anal. Bioanal. Chem., 2005, 381, 317–331 CrossRef CAS.
- X. W. Chen, A. M. Zou, M. L. Chen, J. H. Wang and P. K. Dasgupta, Anal. Chem., 2009, 81, 1291–1296 CrossRef CAS.
- X. B. Long, M. Miró, E. H. Hansen, J. M. Estela and V. Cerdá, Anal. Chem., 2006, 78, 8290–8298 CrossRef CAS.
- S. Latva, S. Peräniemi and M. Ahlgrén, Analyst, 1999, 124, 1105–1108 RSC.
- M. Pena, X. G. Meng, G. P. Korfiatis and C. Y. Jing, Environ. Sci. Technol., 2006, 40, 1257–1262 CrossRef CAS.
- B. J. Lafferty and R. H. Loeppert, Environ. Sci. Technol., 2005, 39, 2120–2127 CrossRef CAS.
- S. Ouvrard, M. O. Simonnot and M. Sardin, Ind. Eng. Chem. Res., 2002, 41, 2785–2791 CrossRef CAS.
- V. Lenoble, C. Laclautre, B. Serpaud, V. Deluchat and J. C. Bollinger, Sci. Total Environ., 2004, 326, 197–207 CrossRef CAS.
- B. L. Batista, D. Grotto, J. L. Rodrigues, V. C. D. Souza and F. Barbosa, Anal. Chim. Acta, 2009, 646, 23–29 CrossRef CAS.
- W. Driehaus, R. Seith and M. Jekel, Water Res., 1995, 29, 297–305 CrossRef CAS.
- G. S. Zhang, J. H. Qu, H. J. Liu, R. P. Liu and G. T. Li, Environ. Sci. Technol., 2007, 41, 4613–4619 CrossRef CAS.
- M. L. Chen, Y. M. Huo and J. H. Wang, Talanta, 2009, 78, 88–93 CrossRef CAS.
- K. S. Subramanian and J. C. Meranger, Anal. Chim. Acta, 1981, 124, 131–142 CrossRef CAS.
- K. Minakata, M. Suzuki and O. Suzuki, Anal. Chim. Acta, 2009, 631, 87–90 CrossRef CAS.
Footnotes |
| † This article is part of a themed issue highlighting outstanding and emerging work in the area of speciation. |
| ‡ Electronic supplementary information (ESI) available: Table S1: Operating parameters of the liquid chromatographic separation-gradient hydride generation-QFAAS detection system. Table S2: The GFAAS temperature program for the determination of arsenic. See DOI: 10.1039/c0ja00091d |
| This journal is © The Royal Society of Chemistry 2011 |