Identification of roxarsone metabolites produced in the system: Soil–chlorinated water–light by using HPLC-ICP-MS/ESI-MS, HPLC-ESI-MS/MS and High Resolution Mass Spectrometry (ESI-TOF-MS)†
Uriel
Arroyo-Abad
ab,
Jürgen
Mattusch
*a,
Monika
Möder
a,
Maria P.
Elizalde-González
b,
Rainer
Wennrich
a and
Frank-Michael
Matysik
c
aUFZ–Helmholtz Centre for Environmental Research, Department of Analytical Chemistry, Permoserstrasse 15, 04318, Leipzig, Germany. E-mail: juergen.mattusch@ufz.de; Fax: +49 341 235 1443; Tel: +49 341 235 1432
bUniversidad Autónoma de Puebla, Centro de Química, Instituto de Ciencias ICUAP, Ciudad Universitaria, Edif. H103, Puebla Pue, 72570, Mexico
cUniversity of Regensburg, Institute of Analytical Chemistry, Chemo-and Biosensors, Universitätsstrasse 31, 93053, Regensburg, Germany
First published on 30th November 2010
Abstract
Roxarsone (4-hydroxy-3-nitrophenylarsonic acid) in contact with soil of volcanic origin and chlorine containing water generated a set of organoarsenicals. The transformation products were identified with element-specific (ICP-MS) as well as molecular-specific (ESI-MS) detection after their HPLC separation. The identification of the main transformation products by means of ESI-MS, ESI-MS/MS and ESI-TOF-MS adduce evidence of chlorinated phenylarsonic acids and a phenylarsine oxide derivative which contains arsenic in the trivalent state. Traces of chlorine in water used for sorption experiments are suggested to be responsible for the formation of chlorinated products. After irradiation of a roxarsone solution with visible light, different transformation product so far not identified were detected.
Introduction
Phenylarsenicals play a key role in the environmental pollution caused by deposited chemical warfare agents1 and animal medication.2 Some phenylarsenicals, specifically, roxarsone (4-hydroxy-3-nitrophenylarsonic acid) and p-arsanilic acid (4-aminophenylarsonic acid), have been widely used for coccidiosis prevention and animal growth promotion in the poultry industry,3–5 as chemotherapeuticals and also as effective growth promoters in the swine industry.6,7Roxarsone and p-arsanilic acid are bioaccumulated in very low concentration and are mainly excreted almost unchanged in manures.5 In poultry litter the concentration of roxarsone ranges from 14 to 54 mg kg−1.8 In manures, also low concentrations of the metabolites: arsenate (AsV), arsenite (AsIII), monomethylarsonic acid, dimethylarsinic acid, 3-amino-4-hydroxyphenylarsonic acid and 4-hydroxyphenylarsonic acid have been detected.9Roxarsone is not naturally occurring in soils, but it is introduced into the soil through the use of poultry litter as fertilizer on cropland.2,10 Garbarino et al. reported that roxarsone is stable in dried poultry litter, but when litter is in contact with water for certain incubation time; arsenate is the main degradation product.11 In studies carried out with manure amended soil, arsenate was the main metabolite of roxarsone, as well.9
Recently we found that phenylarsenicals underwent sorption on volcanic soils from aqueous solution and unknown degradation products were formed after 24 h contact with the soil.12 Due to the fact that roxarsone and its degradation products are highly water-soluble, they propagate into groundwater and can be accumulated by plants.13 In this way, phenylarsenicals and their metabolites can be introduced into the food chain.14–18
In addition, phenylarsenicals can be subjected to transformation owing to microbial activity, increasing the toxicity and mobility of such degradation products. As an example, roxarsone can undergo biotransformation under anaerobic conditions to produce 3-amino-4-hydroxyphenylarsonic acid and inorganic arsenic species as main products.19–21
It is well known that the toxicity of arsenic highly depends on the chemical form in which it is present. It is generally accepted that the trivalent inorganic arsenic compounds are more toxic than the pentavalent one.22 Also the methylated arsenic compounds are harmful and toxic to animals and humans.16,23
Due to the water solubility of the majority of the environmentally related arsenic species, high-performance liquid chromatography (HPLC) and capillary electrophoresis are the preferred separation techniques.24,25 For arsenic speciation analysis, the most popular technique used so far, is HPLC coupled with mass spectrometry (MS). The combination of inductively coupled plasma (ICP) mass spectrometry for elemental identification and electrospray ionization (ESI) mass spectrometry for molecular identification as detector in HPLC is a powerful tool for the identification of unknown arsenic species.24
An increasing number of papers is devoted to the mobility and bioavailability of phenylarsenicals used in animal industry.4,7,20,26 In one of the issues currently under discussion the behaviour of arsenic in cropland fertilized by poultry litter was investigated with respect to irrigation/rain and light exposition.11,17,18
The aim of our studies was to identify metabolites of organoarsenic compounds (especially derived from roxarsone) by suitable combinations of element-specific and molecular-specific mass spectrometric methods. The experiments were performed under simulated environmental conditions by interactions of roxarsone with volcanic soils, chlorinated water and under light irradiation.
Experimental
Materials and methods
For the irradiation experiments a 702 UV-digester (Metrohm, Herisau, Switzerland) equipped with a high pressure mercury lamp HBO 500 was used. The light intensity of this lamp is 2,850 cd and the average light density amounts 30,000 cd cm−2.
Chlorination was performed by bubbling chlorine gas through an aqueous solution of roxarsone. The chlorine gas was produced by oxidation of hydrochloric acid with potassium permanganate.
Analytical methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by a T-piece. The injection volume used was 8 μL.
:1 by a T-piece. The injection volume used was 8 μL.
For identification of the phenylarsonic compounds the ICP-MS peaks at m/z 75 (As) were compared with those peaks obtained by the ESI-MS detector after their separation by means of reversed-phase chromatography (column: Atlantis dC18 (5 μm, 4.6 × 150 mm, Waters, Milford, MA, USA); eluent A: 0.1% HCOOH, 0.1% CH3OH; eluent B: 0.1% HCOOH, 20% CH3OH). The following eluent composition (gradient) was used: 0–3 min 100% A; 3–20 min 25% A (linear); 20–30 min 25% A; 30–31 min 100% A; 31–35 min 100% A. The conditions for the detection are listed in Table 1.
| ICP-MS (Agilent 7500ce) | Conditions |
| RF power | 1500 W |
| Plasma gas flow rate | Ar 15 L min−1 |
| Carrier gas flow | 0.5–0.7 L min−1 |
| Sampling depth | 6–7 mm |
| Ion monitored | m/z 75 (As+) |
| ESI-MS (Agilent MSD 6130) | |
| Polarity | negative |
| Capillary voltage | 4000 V |
| Fragmentor voltage | 200 V |
| Nebulizer pressure | 276 kPa |
| Scan range | 100–300 m/z |
| Spray temperature | 350 °C |
| Nitrogen flow | 11 L min−1 |
| ESI-MS/MS (API 2000) | |
| Polarity | negative |
| Capillary voltage | 5000 V |
| Ion source | Turbolon spray |
| Collision energy | 15 V |
| Product ion scans | molecular ions [M − H]− as precursors |
| Spray temperature | 300 °C |
| ESI-TOF-MS (micrOTOF) | |
| Polarity | negative |
| Capillary voltage | 4000 V |
| End plate offset | 500 V |
| Capillary exit | 150 V |
| Nebulizer gas | (N2): 40 kPa |
| Drying gas | (N2): 4.0 L min−1 |
| Drying temperature | 200 °C |
| Skimmer | 1: −50 V, 2: −23 V |
| Hexapole voltage | 1: −23 V, RF: 100 V |
| Transfer time | 49 μs |
| Pre-pulse storage | 10 μs |
A mixture of propan-2-ol and water (50:50 v/v) containing 0.2% formic acid served as sheath liquid and was pumped with a flow rate of 3 μL min−1 using a syringe pump model KDS 601553 (KD Scientific, Holliston, MA, USA). Sample introduction was performed applying pressure to the sample vial which was connected with the coaxial sheath-liquid sprayer via a short piece of fused silica capillary (50 μm I.D., 360 μm O.D.). As a result, a sample flow through the capillary was generated. The parameters for the operation of the micrOTOF system are also summarized in Table 1.
HPLC: Agilent 1100 with autosampler (Agilent Technologies, Santa Clara, USA); ESI-MS/MS: Triple Quadrupole LC/MS/MS Mass Spectrometer API 2000 (Perkin-Elmer Sciex Instruments, Waltham, Massachusetts, USA). The parameters for detection are presented in Table 1.
Quality control
Blank samples were extracted and analyzed simultaneously with the soil containing samples to determine the potential lability at 25 °C of the roxarsone used in these studies. All experiments were repeated three times and each sample was also analyzed three times.The total concentrations of arsenic in the resulting solutions were determined by ICP-atomic emission spectrometry (CIROS, Spectro A.I., Kleve, Germany).
Results and discussion
HPLC-ICP-MS/ESI-MS
For the separation of roxarsone and its transformation products reversed phase chromatography was chosen using gradient elution with methanol. The element-specific and molecular-specific detection was performed as described above by parallel coupling ICP-MS and ESI-MS. Overlaying the ICP-MS chromatogram with that of the ESI-MS facilitates the molecular mass search for arsenic containing compounds. In Fig. 1 the chromatograms of a freshly prepared roxarsone solution (ROX) in comparison to roxarsone treated 24 h by three different soils (AT, LO, TL) employing ICP-MS detection are presented. When roxarsone was in contact with the soils, additional peaks in the arsenic selective chromatograms could be detected indicating that besides sorption on the soil, also more than 10 transformation products were formed during the experiment. They were produced in different amounts as indicated by the peak heights/areas in the chromatograms detected by ICP-MS. The highest degree of transformation of roxarsone was observed using the volcanic soil TL. Because of the limited sensitivity of ESI-MS detection, the identification was focused on the major arsenic containing transformation products at retention times 6.3 (peak A), 16.2 (peak B) and 24.5 (peak C) minutes. They could be identified by their molecular mass peak in the scan - mode spectra using negative ionization (Fig. 2). Thereby peak A corresponds to m/z 289, peak B to m/z 296 and peak C to m/z 280 (each as [M − H]−). In the case of peaks B and C the isotopic pattern indicates the presence of chlorine in the molecule, which can be observed for the molecular mass peak as well as for the fragments.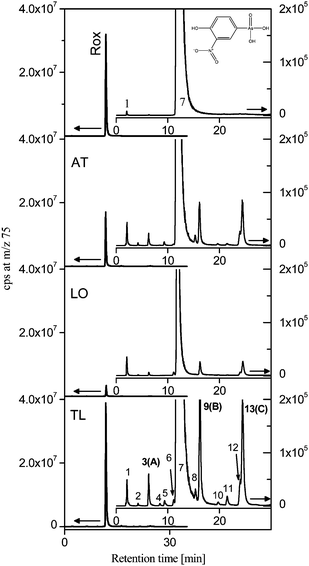 | ||
| Fig. 1 HPLC-ICP-MS chromatograms of the initial solution of roxarsone (ROX) and solutions containing soluble transformation products detected after 24 h contact with the Acrisol (AT), Andosol (LO) and Tepetate (TL) soils: Left axis – full scale chromatograms; right axis – zoomed chromatograms. | ||
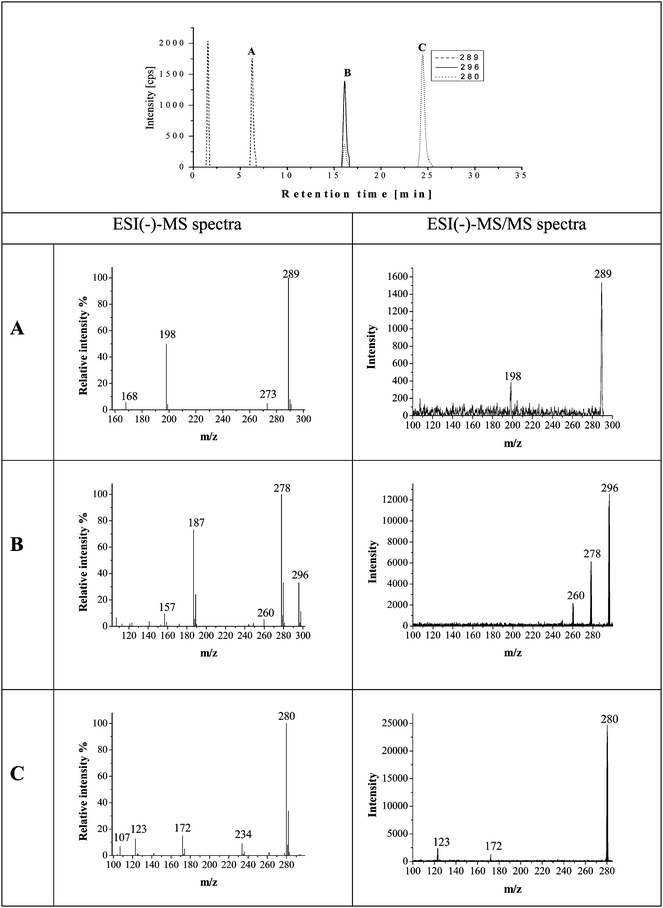 | ||
| Fig. 2 HPLC-ESI-MS chromatograms in SIM mode on m/z 280, 289 and 296, and negative ESI-MS and ESI-MS/MS spectra of the peaks in the scan mode (m/z 100–300) of roxarsone solution in contact with the TL soil. | ||
HR ESI-TOF-MS
To determine the exact molecular masses of the unknown arsenic containing components, the solution carrying the transformation products of roxarsone was injected directly in an ESI-TOF-MS without chromatographic pre-separation. Fig. 3 shows the resulting mass spectra in the range of 275 to 300 m/z. The m/z – values in the high resolution mode agree with the results obtained by ESI-MS and ESI-MS/MS (Tab. 2). The differences (Δm) between the theoretical masses and the measured ones of the identified compounds A, B and C vary within 0.7 and 2.1, which is an acceptable value (< 3 ppm) for those measurements. Also the isotopic pattern of the sample compared with the simulated isotopic pattern (SIP) for peaks B and C, respectively, proved that these both compounds are chlorine-containing phenylarsonic acid derivatives as illustrated in Fig. 4.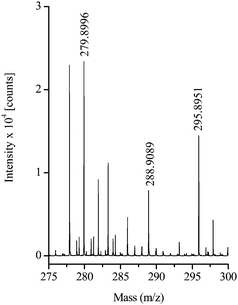 | ||
| Fig. 3 ESI(-)-TOF-MS spectrum of the roxarsone solution after contact with the Tepetate soil. | ||
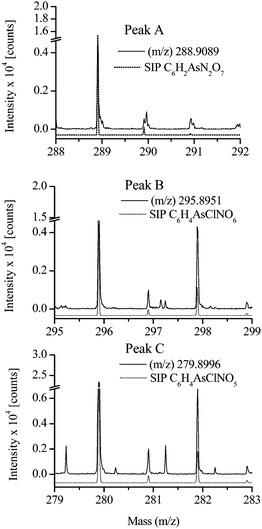 | ||
| Fig. 4 Comparison of the ESI(-)-TOF-MS spectra with simulated isotopic pattern (SIP). | ||
HPLC-ESI-MS/MS
For a positive proof of the structure of the transformation products the findings by HPLC-ICP-MS/ESI-MS and HR ESI-TOF-MS have to be supported by HPLC-ESI-MS/MS studies. These investigations were performed in the negative ionization mode. The precursor ions [M − H]− = 280, [M − H]− = 289 and [M − H]− = 296 were analyzed by product ion scans. In general, the resulting product ions generated by collision activated dissociation (CAD) showed a good conformity with those one detected in the spectra using the scan mode ESI(-)-MS shown in Fig. 2.For peak A, the precursor ion at m/z 289 was fragmented in one main product ion at m/z 198 (A2). The other fragments at m/z 273 (A4) and 168 (A3) detected by ESI(-)-MS could not be confirmed by ESI-MS/MS because of its lower sensitivity. For peak B, the precursor ion at m/z 296 was fragmented in two main product ions at m/z 278 and 260. The first fragment was also observed in the ESI(-)-MS studies with high intensity. The second one shows only a low abundance in the ESI(-)-MS spectra without an isotopic pattern of chlorine. The precursor ion at m/z 280 (peak C) was fragmented in two small product ions corresponding to m/z 172 and 123 corresponding to AsO3− shown in spectrum C in Fig. 2. It is not to be excluded that a co-eluting compound can generate this fragment. However, the simultaneous occurrence of m/z 107 for AsO2− and 123 for AsO3− additional to m/z 75 obtained by ICP-MS detection can be regarded as indication for the real existence of this fragments. The fragment m/z 234 was only detected by ESI(-)-MS as a chlorine containing one.
The compound indicated as peak B is proposed to be the anion of 3-chloro-4-hydroxy-5-nitrophenylarsonate based on the molecular mass at m/z 296 (B1). By loss of water (Δ m/z 18) the main fragment 3-chloro-4-hydroxy-5-nitrophenylarsenite with m/z 278 (B2) was formed that is further fragmented to the radical anion 2-chloro-6-nitro-4-oxocyclohexa-2,5-dienolate fragment with m/z 187 (B3, Δ m/z 91) by cleavage of AsO. The elimination of oxygen results in 2-chloro-6-nitrophenolatem/z 157 (B4, Δ m/z 15). Alternatively, the fragment 4-hydroxy-5-nitrophenylarsenite (B5 m/z 260) derived from B2 was formed by substitution of chlorine by a hydroxyl group on the aromatic ring.
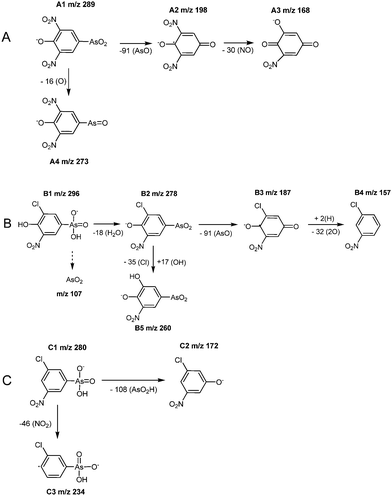 | ||
| Fig. 5 Schemes proposed for fragmentation pathways to identify the peaks A, B and C in Fig. 2. | ||
In the case of peak C, the proposed compound is 3-chloro-5-nitrophenylarsonic acid based on the molecular mass at m/z 280 (3-chloro-5-nitrophenylarsonate, C1). The loss of the arsonate group (AsO2H) results in 3-chloro-5-nitrophenolate at m/z 172 (C2, Δ m/z 108). Considering an alternative pathway the anionic radical fragment 3-chlorophenylarsonate at m/z 234 (C3, Δ m/z 46) was detected, which can be attributed to the lost of the nitro group.
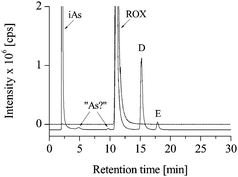 | ||
| Fig. 6 HPLC-ICP-MS chromatograms of untreated (dotted line) and chlorine-treated (solid line) roxarsone solutions; (iAs) inorganic arsenic. | ||
After longer treatment, roxarsone was degraded completely, and a couple of new arsenic containing compounds in the retention time window of 2 to 5 min appeared. Further experiments are necessary to identify these products after chlorine treatment.
The observed degradation and chlorination reactions created numerous new arsenic species with unknown toxicity. The agreement of only one transformation product in the chlorination experiments with that in the soil experiments shows that the reaction pathways are to be governed by other factors like catalytic activities of the soils. Disinfections by chlorine or hypochlorite are standard methods for waste water, tap water treatment and cleaning of buildings for livestock. Because of their global application it can also come in contact with the feed additives roxarsone or arsanilic acid that not only the danger exist that more toxic inorganic arsenic is formed but also organic derivatives of the stock compounds.
Conclusion
The treatment of roxarsone in aqueous suspensions of volcanic soils leads to numerous transformation and degradation products, which contain arsenic as heteroatom. The identification by means of elemental and molecular-specific detection can be effectively supported by complementary approaches, like ESI-MS/MS and ESI-TOF-MS.One of the transformation products formed in contact with soils was identified as arsenic(III) containing compound, whereas the other both are chlorinated organoarsenicals. One of these compounds (3-chloro-4-hydroxy-5-nitrophenylarsonate) could be synthesized by direct chlorination of roxarsone. With intensive UV - irradiation of roxarsone in solution a fast degradation to inorganic arsenic species occurs in contrast to treatment under visible light. In the second case numerous organoarsenicals in low concentrations were formed. Future studies are in progress to identify the degradation and transformation products formed and to clarify their reaction pathways and toxicity.
Acknowledgements
Financial support of Uriel Arroyo-Abad (MSc) was granted through CONACyT (Mexico) and Helmholtz Association (Germany).References
- M. Leermarkers, W. Baeyens, M. De Gieter, B. Smedts, C. Meert, H. C. De Bisschop, R. Morabito and P. Quevauviller, TrAC, Trends Anal. Chem., 2006, 25, 1–10 CrossRef CAS.
- B. P. Jackson and P. M. Bertsch, Environ. Sci. Technol., 2001, 35, 4868–4873 CrossRef CAS.
- A. K. Sarmah, M. T. Meyer and A. B. A. Boxall, Chemosphere, 2006, 65, 725–759 CrossRef CAS.
- K. C. Makris, J. Salazar, S. Quazi, S. S. Andra, D. Sarkar, S. B. H. Bach and R. Datta, J. Environ. Qual., 2008, 37, 963–971 CrossRef CAS.
- J. L. Morrison, J. Agric. Food Chem., 1969, 17, 1288–1290 CrossRef CAS.
- L.-F. Hu, H.-J. Feng, Y.-Y. Wu, Y.-Y. Long, J. Wan and D. S. Shen, J. Hazard. Mater., 2010, 174, 194–201 CrossRef CAS.
- K. C. Makris, S. Quazi, P. Punamiya, D. Sarkar and R. Datta, J. Environ. Qual., 2008, 37, 1626–1633 CrossRef CAS.
- R. Sierra-Alvarez, I. Cortinas and J. A. Field, J. Hazard. Mater., 2010, 175, 352–358 CrossRef CAS.
- L. Yao, G. Li, Z. Dang, Z. He, C. Zhou and B. Yang, Plant Soil, 2009, 316, 117–124 CrossRef CAS.
- B. L. Brown, A. D. Slaughter and M. E. Schreiber, Appl. Geochem., 2005, 20, 123–133 CrossRef CAS.
- J. R. Garbarino, A. J. Bednar, D. W. Rutherford, R. S. Beyer and R. L. Wershaw, Environ. Sci. Technol., 2003, 37, 1509–1514 CrossRef CAS.
- U. Arroyo-Abad, M. P. Elizalde-González, C. M. Hidalgo-Moreno, J. Mattusch and R. Wennrich, J. Hazard. Mater., submitted Search PubMed.
- L.-X. Yao, G.-L. Li, Z. Dang, Z.-H. He, C.-M. Zhou and B.-M. Yang, J. Hazard. Mater., 2009, 164, 904–910 CrossRef CAS.
- C.-W. Liu, C.-C. Lin, C.-S. Jang, G.-R. Sheu and L. Tsui, J. Plant Nutr. Soil Sci., 2009, 172, 550–556 CrossRef CAS.
- A.-C. Schmidt, K. Kutschera, J. Mattusch and M. Otto, Chemosphere, 2008, 73, 1781–1787 CrossRef CAS.
- L. X. Yao, G. L. Li, Z. Dang, Z. H. He, C. M. Zhou and B. M. Yang, Geoderma, 2009, 154, 48–51 CrossRef CAS.
- A. J. Bednar, J. R. Garbarino, I. Ferrer, D. W. Rutherford, R. L. Wershaw, J. F. Ranville and T. R. Wildeman, Sci. Total Environ., 2003, 302, 237–245 CrossRef CAS.
- D. W. Rutherford, A. J. Bednar, J. R. Garbarino, R. Needham, K. W. Staver and R. L. Wershaw, Environ. Sci. Technol., 2003, 37, 1515–1520 CrossRef CAS.
- P. Chovanec, J. F. Stolz and P. Basu, Metallomics, 2010, 2, 133–139 RSC.
- J. F. Stolz, E. Perera, B. Kilonzo, B. Kail, B. Crable, E. Fisher, M. Ranganathan, L. Wormer and P. Basu, Environ. Sci. Technol., 2007, 41, 818–823 CrossRef CAS.
- I. Cortinas, J. A. Field, M. Kopplin, J. R. Garbarino, A. J. Gandolfi and R. Sierra-Alvarez, Environ. Sci. Technol., 2006, 40, 2951–2957 CrossRef CAS.
- M. F. Hughes, Toxicol. Lett., 2002, 133, 1–16 CrossRef CAS.
- E. M. Kenyon and M. F. Hughes, Toxicology, 2001, 160, 227–236 CrossRef CAS.
- J. Mattusch, D. Möller, M. P. Elizalde González and R. Wennrich, Anal. Bioanal. Chem., 2008, 390, 1707–1715 CrossRef CAS.
- K. Kutschera, A.-C. Schmidt, S. Köhler and M. Otto, Electrophoresis, 2007, 28, 3466–3476 CrossRef CAS.
- E. K. Silbergeld and K. Nachmann, Ann. N. Y. Acad. Sci., 2008, 1140, 346–357 CrossRef CAS.
- J. D. Etchevers, C. Hidalgo, C. Prat and P. Quantin, in Encyclopedia of Soil Science, ed. R. Lal, Marcel Dekker Inc., New York, 2006, pp. 1745–1748 Search PubMed.
- C. Hidalgo, J. D. Etchevers, A. Martínez-Richa, H. Yee-Madeira, H. Calderon, R.Vera-Graziano and F. Matus, Appl. Clay Sci., 2010, 49, 348–358 CrossRef CAS.
- B. Prado, C. Duwig, C. Hidalgo, D. Gómez, H. Yee, C. Prat, M. Esteves and J. D. Etchevers, Geoderma, 2007, 139, 300–313 CrossRef CAS.
- C. D. Murphy, J. Appl. Microbiol., 2003, 94, 539–548 CrossRef CAS.
- R. Ullrich and M. Hofrichter, FEBS Lett., 2005, 579, 6247–6250 CrossRef CAS.
Footnote |
| † This article is part of a themed issue highlighting outstanding and emerging work in the area of speciation. |
| This journal is © The Royal Society of Chemistry 2011 |