Contribution to selenium speciation in cerebrospinal fluid samples†
Bernhard
Michalke
*a and
Achim
Berthele
b
aHelmholtz Center Munich–German Research Center for Environmental Health, Ingolstädter Landstr. 1, 85764, Neuherberg, Germany. E-mail: bernhard.michalke@helmholtz-muenchen.de
bDepartment of Neurology, Klinikum rechts der Isar, Technische Universität München, 81675, Munich, Germany
First published on 26th November 2010
Abstract
The well known beneficial health effects of Se have demanded the development of speciation methods for providing deeper insight into Se-metabolism and transport. This is also of significant importance for healthy brain function. Thus, Se-speciation was performed in 15 individual cerebrospinal fluid (CSF) samples (a′3 replicates) from neurologically healthy persons. First size exclusion chromatography (SEC) coupled to inductively coupled plasma mass spectrometry (ICP-MS) equipped with a dynamic reaction cell (DRC) was used for monitoring the most abundant 80Se isotope. SEC separation provided Se-species characterization distinguishing between seleno-proteins and Se- low molecular mass (LMM) compounds. However, for improved speciation information a method based on strong anion exchange (SAX) separation was employed and optimized for CSF samples. The analysis of CSF samples from different individuals revealed six Se-species comprising relative mean concentrations of 8.5% (range: 5–11%), 27% (range: 20–31%), 2.2% (1.7–3.9%), 17% (range: 11–19%), 26% (range: 24–31%) and 20% (range: 19–21%) of the total Se amount, which in average was 3.6 μg L−1 (range: 2.9–4.8 μg L−1). Single and mixed Se-standard compounds were analyzed for verifying respective retention times, using serial UV- and ICP-MS detection. Additionally, standard additions were made to CSF samples for further peak identifications. By means of standard matching selenate, thioredoxinreductase (TrxR) and Se-albumin (Se-HSA) were found. No matches for Se methionine (SeM), Se-cystine (SeC), glutathioneperoxidase (GPx) and selenite were seen. Since no standard for selenoprotein P (SePP) was commercially available, this compound was prepared freshly from human serum using a Heparin-affinity column. It turned out that this SePP fraction showed matched retention time with the first peak in SAX separation, but also with selenate when age of the prepared standard was increasing.
Introduction
Selenium and the central nervous system and CSF
Selenium is an essential trace element for humans. Its beneficial role for human health is generally accepted and includes protection against oxidative stress, prevention of heart diseases and cancer.1,2. Also thyroid metabolism is dependent on Se as deiodinases are seleno-proteins.3 In the central nervous system, oxidative stress has been implicated in pathophysiology of neurodegenerative diseases, such as Alzheimer′s (AD) and Parkinson′s disease (PD) and even stroke.4–7 Fahn and Cohen8 published data suggesting oxidative stress specifically in the substantia nigra of PD patients. Protection against oxidative stress is based on superoxide dismutase activity, and in a subsequent step on the activity of the seleno-enzymes glutathioneperoxidase and TrxR, being present in brain.4 Brain Se levels are primarily maintained by selenoprotein P (SePP) directly expressed in neuronal tissue,9,10 which in turn depends on proper Se supply across the blood brain barrier (BBB). Cerebrospinal fluid (CSF) is mainly an excretion of the choroid plexus in the brain ventricles. It plays an important role in the homeostasis and metabolism of the central nervous system.11 Since CSF has close molecular exchange with the extracellular space of brain parenchyma the compositions of CSF and extracellular brain fluids are similar and a misbalance, a depletion of elements or change of element species in the brain is likely to be reflected in CSF. Se depletion or changes in Se speciation in neurological diseases can be monitored in the CSF. Total Se concentration in CSF was described to be low in healthy control subjects (approx. 1.2 μg L−1–4 μg L−1) in recent papers which adhere to sufficient quality control (QC) measures12–14 or around 13 μg L−1 in papers before 2000 where no QC were clearly described.7,15Se-Speciation in CSF samples
Open questions arise about the transport of Se species to the brain across the neural barriers.14 In previous work we have shown that the total Se concentration in CSF is independent from serum selenium concentration and a strictly controlled and regulated pathway across BBB was assumed. For understanding how and in which form selenium is crossing the neural barriers and enters the brain, it is necessary to identify the Se species in CSF. Although Se-speciation is required for understanding this mechanism no such data were found when screening ISI web-of knowledge or PubMed, except for single Se-species like Selenoprotein P (SePP). SePP was found to be transcripted from brain independently from liver-borne SePP in mice.9In the present paper, analysis of Se species in CSF of 15 individuals (a′3 replicates) was performed first with SEC coupled to ICP-DRC-MS, and in the main approach with SAX-ICP-DRC-MS. The DRC mode of the mass spectrometer was chosen for achieving highest sensitivity by monitoring the main Se isotope 80Se. In accordance with the manufacturer′s instruction manual and Jitmanee et al.16 methane was used as the DRC gas.
A previously developed SAX method from Xu et al.17 was used for separation of SePP, GPx and Se-albumin (HSA). The elutions of further seleno compounds such as thioredoxin reductase (TrxR), Se-methionine (SeM), Se-cystine (SeC), selenite and selenate were additionally checked.
With this method, Se speciation was analyzed in CSF samples from neurologically healthy humans as a first approach to gain information about Se-species behind the blood brain barrier.
Experimental
Chemicals and reagents
Chemicals and reagents used throughout this work were of suprapure grade. Certified selenium and rhodium stock standards (1000 mg L−1) were purchased from CPI (Santa Rosa, USA). Selenite and selenate, Se-methionine, Se-cystine, TrxR from rat liver (EC 1.8.1.9.), glutathioneperoxidase from bovine erytrhorzytes (EC 232-749-6), human serum albumin (HSA) and TRIS buffer were purchased from Sigma-Aldrich, Deisenhofen, Germany. Ammonium acetate and acetic acid were bought from Merck, Darmstadt, Germany. Arliq and methane (99.999% purity) were purchased from Air Liquide, Gröbenzell, Germany.Selenite and selenate stock solutions were prepared at a concentration of 1000 mg Se/L by appropriate weighing the substances and dissolving in Mili-Q water (18.2 MΩ cm) from a Milli-Q system (Millipore, Bedford, MA, USA). HSA was prepared accordingly at a concentration of 1000 mg L−1. Preparation of a Se-HSA was done by mixing 10 mg Se/L selenite to this stock solution and incubation for at least 14 days.
Working standards of Se-species were prepared daily from their stock standard solutions by appropriate dilution with Milli-Q H2O.
For quality control during total Se determination human serum and urine from Recipe, Munich, were reconstituted as indicated on the respective flask labels. The resulting solutions were diluted 1/50 (serum) and 1/10 (urine) with Milli-Q water before measurements. The manufacturer′s target mean values (62 μg L−1 for serum and 23 μg L−1 for urine) were found (60 ± 3.2 μg L−1 or 24 ± 2.1 μg L−1).
SEC parameters
The size characterization of Se species from CSF was performed using the Knauer 1100 Smartline inert Series gradient HPLC system connected to two serial SEC columns to provide separation of proteins (150 × 8 mm, TSK 55 F gel) as well as improved separation of LMM compounds (300 × 8 mm, TSK 40 S gel). The eluent (Tris-HCl, 10 mM, pH 7.4) was pumped isocratically at a flow rate of 0.7 ml min−1. The sample volume was 100 μL. The column effluent was directed first to an auxiliary UV detector which was installed directly before the nebulizer at ICP-MS. UV was detected at 220 nm. For checking a possible sticking of Se compounds, the column was purged after each run with modified eluent, where 50 mM NaCl were added and pH was set to 2. No additional Se peaks were monitored during purging.For estimating elution times, a simple mass calibration was performed with standard solutions of GPx, HSA, SeC and selenate. The proteins eluted around 9.6 min and SeC or selenate at 15.7–16 min.
SAX-Parameters
SAX separation was done accordingly to Xu et al.17 using the Knauer 1100 Smartline inert Series gradient HPLC system connected to an anion exchange column ProPac SAX-10 (250 × 2 mm I.D.). The sample volume was 100 μL. The mobile phases were: eluent A: 10 mM Tris-HAc buffer, pH 8.0; and eluent B: A + 500 mM ammonium acetate, pH 8.0. For optimized separation of Se-species from CSF the gradient was flattened and was finally: 0–3 min 100% eluent A; 3–10 min 100–60% eluent A, (0–40% eluent B); 10–20 min 60–0% eluent A (40–100% eluent B); 20–35 min 100% eluent B, 35–42 min 100% eluent A. The flow rate was 0.23 mL min−1. The column effluent was mixed with 1 μg L−1 Rh (final concentration) as internal standard (total flow rate: 0.3 mL min−1) and directed to ICP-MS.Preparation of SePP from human serum as a standard
Because SePP was not commercially available, it was purified from human plasma using affinity chromatography, based on Shigeta et al.18 and slightly modified according to the column instruction sheet. Briefly 200 μL serum were injected on a Heparin-affinity column (Amersham, GE Healthcare Europe GmbH, München, Germany) and chromatographed using a linear gradient of A = 50 mM Tris, 10 mM NH4-acetate/acetic acid, pH 6.0 to buffer B = A but 800 mM NH4-acetate, pH 8.5, within 12 min and remaining at 100% B for further 8 min. SePP was eluted at a flow rate of 1.0 mL min−1 and collected under 280 nm monitoring. Se was subsequently determined in fractions with FI-ICP-DRC-MS.14 The SePP fraction was preconcentrated by freeze drying and was re-dissolved in 1 mL of 10 mM Tris-HCl buffer, pH 7.2. The resulting SePP solution was stored at −20 °C until use.Inductively coupled plasma mass spectrometry
Table 1 shows the experimental settings chosen for ICP-DRC-MS after optimization.| Instrument | Perkin Elmer Sciex ELAN DRC II, Toronto, Canada |
|---|---|
| Plasma conditions | |
| Rf power/W | 1250 |
| Plasma gas flow/L min−1 | 15 |
| Auxiliary gas flow/L min−1 | 1.05 |
| Nebulizer gas flow/L min−1 | 0.83, daily optimized |
| Mass spectrometer settings | |
| Dwell time/ms | 300 |
| Sweeps per reading | 6 |
| Readings per replicate | 1000 |
| Autolens | On |
| Ions monitored | 80Se, 32S, 103Rh |
| Reaction gas | CH4 |
| Reaction gas flow rate/ml min−1 | 0.6 |
| Rejection parameter q | 0.6 |
| Rejection parameter a | 0.0 |
Total Se determination in CSF and peak quantification from chromatograms
Flow injection analysis ICP-DRC-MS was applied for total Se determination in CSF according to our previously published method.14 In short terms: A Knauer 1100 Smartline inert Series HPLC system equipped with a binary pump, a vacuum degasser and an electronic valve with a 25μl injection loop (Perkin Elmer, Rodgau-Jügesheim, Germany) was directly coupled to ICP-DRC-MS. The flow rate was 1 ml min−1 of 50% eluent A and B, each, from SAX separation. CSF-samples were diluted 1: 1.1 by adding 1 μg L−1 Rh (final conc.) as internal standard.Peak quantification from chromatograms was done by comparing peak areas with peak area calibration curves from FI-ICP-DRC-MS.
Data processing of FI-ICP-(DRC)-MS
Rh and Se data files were exported from the ELAN software and processed with the Knauer HPLC software “Clarity” for peak area integration. For each sample (or standard) a quotient of Se-peak area to Rh-peak area was calculated and taken as the result corrected for the internal standard (Rh). These values were used for the calibration curve (standards) or for calculating the concentration according to the calibration curve (samples).
Results and discussion
SEC-ICP-DRC-MS
As a first screening and characterization of Se-species in CSF a SEC-ICP-DRC-MS analysis was conducted. Since it is generally accepted that the blood brain barrier favours passing small molecules more than (bigger) proteins, a combination of two SEC columns was used to provide separation of proteins (150×8 mm, TSK 55 F gel) as well as improved separation of LMM compounds (300×8 mm, TSK 40 S gel). Contrary to the above expectation to find LMM-Se species, mainly HMM Se compounds were seen. A sharp peak for seleno-proteins was monitored accounting for 95% of Se eluting from CSF, whilst only a very small single peak signal of LMM Se species appeared amounting to about 5% of eluting Se. Peakshapes of the two peak signals were each looking as either from single or from coeluting compounds. A purging step similarly to Nischwitz et al.19 using the chromatography eluent but with 50 mM NaCl at pH 2 revealed no further Se-compounds, proving that no loss (sticking to column) of Se-species during chromatography appeared. Subsequent improved approaches using SEC separation optimized for protein separation did not even sufficiently resolve CSF–Se species. Only broad and unresolved Se signals with peak heights being close to noise were monitored, eluting at retention times being typical for a mass range of ca. 110–50 kDa. The worse signal-to noise ratio might be explained by the low total Se concentration (and consequently low Se species concentration) combined with the analyte dilution during SEC separation.20 The insufficient resolution may be due to the partly similar molecular masses of investigated Se-species, where e.g. GPx is assigned to 83 kDa,21 HSA to 66 kDa22 and SePP between 50 and 65 kDa (depending on determination method).23,24SAX-ICP-DRC-MS
Therefore another attempt was performed, based on the method from Xu et al.17 developed for Se-speciation in human serum. Xu′s method uses SAX-ICP-MS. After having installed the method, operation and elution was first checked by running a laboratory reference serum. Elution of three Se peaks was observed (not shown) in close coincidence with chromatograms from the paper by Xu, where these three peaks were asigned to as SePP, GPx and Se-HSA. As a next step, preliminary separations of CSF samples were conducted to elucidate whether the method also fits to these kinds of samples. There, several Se species eluted between 15–20 min which were still insufficiently resolved. Therefore the chromatographic elution was optimized for CSF samples by a slight flatening of the gradient (cf. experimental) which resulted also in changed retention times for SePP (2.91 min), GPx (17.12) and Se-HSA (20.40 min). These retention times were checked by standard compounds and by separating the reference serum.With this optimized method the subsequent analysis of CSF samples and mixtures of Se- standard compounds for peak identification were performed. Fig. 1 shows typical chromatograms of three CSF samples as examples. Each of the chromatograms has six distinct Se-peaks at elution times of 2.91, 18.02, 19.48, 20.41, 27.45 and 33.70 min. These peaks comprised to (mean ± SD) 0.31 ± 0.14 μg Se/L (mean 8.1%), 1.1 ± 0.4 μg Se/L (mean 28.6%), 0.1 ± 0.04 μg Se/L (mean 2.6%), 0.61 ± 0.14 μg Se/L (mean 15.9%), 1.0 ± 0.16 μg Se/L (mean 26%), and 0.72 ± 0.1 μg Se/L (mean 18.8%), of the total eluting Se amount, which was 3.84 ± 0.59 μg Se/L. The mass balance (summed peaks/total Se) mounted to 105 ± 9%.
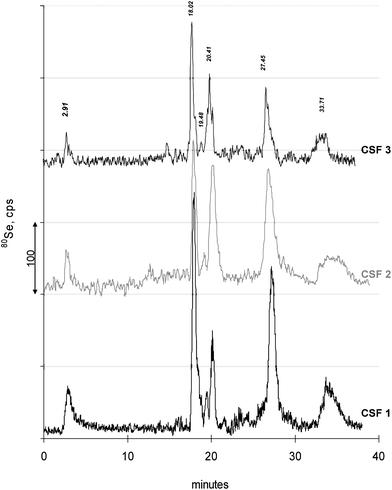 | ||
| Fig. 1 Examples of typical chromatograms from three individual CSF samples are seen. In total, samples from 15 individuals (a′3 replicates) were analyzed. Replicate measurements resulted in equal chromatograms. Elution times of Se-peaks are given representatively for the chromatogram CSF 3 and differed (if ever) insignificantly for the other chromatograms. | ||
Subsequently, single and mixed Se-standard compounds were analyzed for verifying respective retention times, using serial UV- and ICP-MS detection, the latter monitoring 80Se (and 16S isotopes, not shown in the figure). Fig. 2 shows a representative CSF sample (line A) compared to the chromatograms of several standard compounds (Fig. 2, lines B-D), either SeM, GPx, TrxR, Se-HSA, selenite and selenate (Fig. 2, line B) or SeM, SeC, selenite and selenate (Fig. 2, line C). It can be seen that TrxR, Se-HSA and selenate showed matched retention times with peaks at 19.48, 20.41 and 27.45 min. The match was further confirmed when (single-) standard additions of these compounds to CSF samples were performed (not shown). However, no retention time match was observed for standards (even not after standard addition to CSF sample) of SeM, SeC, GPx and selenite. Therefore, the presence of these Se-compounds in CSF could not be confirmed. An important observation was the variation in stability of the diluted (untreated) GPx standard when being chromatographed. The peak area reproducibility of the GPx standard was low and additional selenite and an even higher selenate peak appeared, the more the GPx peak at 17.12 min disappeared. After addition of selenite to the GPx standard solution, a sufficient reproducibility of GPx peak area at 17.12 min was observed. Apparently, the stability of the compound was low at low Se concentration.
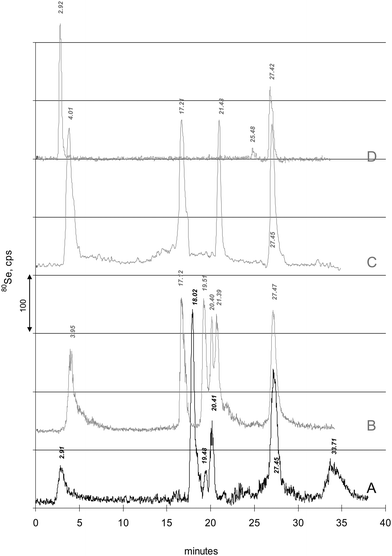 | ||
| Fig. 2 Retention time match of peaks is shown. For comparison and peak matching or mismatching, the typical chromatogram of a CSF sample “A” is plotted. Elution times of Se-peaks are indicated at peak maxima. Grey lines: “B”: Chromatogram of SeM (3.95 min), GPx (17.12 min), TrxR (19.51 min), Se-HSA (20.40 min), selenite (21.39 min) and selenate (27.47 min). Se concentration was 1 μg Se/L each (for HSA/selenite mixture 1 μg L−1 selenite). A retention time match is observed for TrxR, Se-HSA and selenate. For TrxR and Se-HSA retention time was also confirmed by UV detection and 32S monitoring (not shown). “C”: Chromatogram of SeM (3.95 min), SeC (17.21 min), selenite (21.43 min) and selenate (27.40 min), each 1.5 μg Se/L. Retention time match is observed only for selenate. “D”: Chromatogram of SePP and selenate (27.40 min). SePP was prepared from human serum using Heparin-affinity chromatography. Retention time match is observed for both SePP and selenate. Aging of the SePP fraction at 4 °C resulted in a decreased SePP peak but an increase of selenate. | ||
Although, the first peak at 2.91 min eluted at the same time as the one from serum being asigned as SePP in the paper of Xu, an additional verification was essential. As no SePP standard was commercially available, SePP was prepared using a Heparin-affinity column. Fig. 2, line D shows the comparison of the SAX-ICP-MS chromatogram of this SePP fraction to the CSF sample (Fig. 2, line A). It gets obvious that the peak at 2.91 min from CSF coelutes with the SePP standard prepared from serum, but also selenate was seen. It should be noted that reproducing the SePP chromatogram immediately after the first run (SePP fraction standing at 4 °C in the refrigerator during the 40 min chromatographic separation of the first run) resulted in a decrease of SePP peak paralleled by an analogous increase of selenate. The third replicate (SePP fraction standing 80 min in the refrigerator) revealed no SePP but solely selenate. However, after a second Heparin-affinity preparation, aliquotation and immediate deep freezing of each aliquot until use, the prepared SePP standard showed longer stability. No significant difference between the first and second run was seen as each SePP aliquot was freshly thawed. A fast decomposition of SePP was also observed from Ulrich Schweizer (pers. comm.). This tendency for decomposition of SePP was taken first into consideration as an explanation for the contradiction between our first results from SEC- compared to SAX-ICP-DRC-MS: Selenate is eluting at around 15.85 min during SEC separation, where only 5% of total Se was found from CSF. Contrary, selenate amounted to 26% of total Se during SAX separation. However, the rapid loss of SePP during 4 °C storage seemed not to be causative, since a) CSF samples were frozen within minutes after drawing and kept entirely frozen until analysis, and b) this could not explain the different amounts of selenate/LMM compounds between SAX and SEC separations, each analysed directly after thawing. To clarify this issue both experiments (SEC and SAX) were repeated in turned sequence which did not influence the results. Besides, in a former, preliminary approach, Schweizer and co-workers (Ulrich Schweizer, J. Köhrle, B. Michalke, unpublished results) performed a targeted analysis on SePP in CSF using an assay developed by Hollenbach et al.25 They found a mean concentration of 15 μg L−1 SePP (as whole protein) in CSF, which contributes only to about 7% of total Se in CSF. This low contribution is in acceptable coincidence with our finding of ca. 8.5% from total Se in CSF.
Although eight Se-species were used for standard matching, the peaks at 18.02 and 33.70 min remained unidentified. Additional investigations with ESI-MS unfortunately were not successful due to the very low species concentrations below LoD of the instrument and the SAX-chromatographic eluents being not very suitable for ES ionization.
The finding of Se-HSA in CSF was not surprising. It is known, that HSA is crossing BBB in small extent and the HSA-quotient of CSF/serum is the standard mean to evaluate the intactness of the barrier. Since Se-HSA is generally accepted for serum e.g.17,24,26,27 Se-HSA aside from HSA is transported in some amount across BBB into CSF. Jitaru et al.27 presented Se-speciation in serum where Se-HSA amounted to about 22% of total Se (19 ng ml−1 Se-HSA of 86 ng ml−1 total Se). The presence of SePP in CSF should also be expected. SePP is even smaller than HSA. However, independent from SePP in serum, Scharpf et al.9 found SePP being locally expressed in the brain and both, SePP and SePP-mRNA were found in human CSF. Further, SePP seems to play an important role in neuronal survival by protection against reactive oxygen species (ROS).28 The presence of TrxR fits well to the supposed protective action against ROS.29 The primary defense line against oxidative stress is based on superoxide dismutase activity, which however, is resulting in H2O2 production. Subsequently peroxides are eliminated by the activity of the seleno-enzymes TrxR and GPx, both being known in brain.4 Arnér et al.,29 too, had found GPx in brain. Kühbacher et al.30 analyzed brain samples from Se deficient and Se supplemented mice and found glutathioneperoxidase specifically in those brain regions, which are in molecular exchange with ventricles and thus also with CSF. Walther et al.12 and Pyne-Geithman et al.31 analyzed GPx in CSF. Therefore, it was surprising that we could not find GPx in CSF. We carefully re-evaluated all data, specifically those of GPx addition to CSF. But this, too, confirmed that the GPx addition peak appeared at 17.12 min (in all cases clearly before the high non-identified peak) and did not match a detectable native peak from CSF. Furthermore, GPx from our reference serum appeared at 17.12 min retention time too, proving that GPx in biological matrix can be found at this elution time.
The observed chromatographic instability of GPx in the low μg Se/L range (see above) could be an explanation for both, the (seemingly) absence of GPx in CSF and for the contradiction between SEC- and SAX-ICP-DRC-MS. In analogy to the non-stabilized standard, CSF-GPx could have been degraded during SAX-separation, thus disappearing at its specific retention time (17.12 min) and mounting up as selenate. This assumption would explain both, the missing of GPx in CSF and the elevated selenate amount after SAX separation (but not after SEC separation). Temperature and chemically induced reduction of GPx stability has been observed previously,32–34 but presently, these findings are not completely explained and need further investigation.
Summary
We investigated CSF samples from neurological healthy persons and conducted Se speciation investigations. SEC-ICP-DRC-MS revealed the main part of Se being associated to proteins, and only about 5% may be attributed to LMM compounds or inorganic Se. However, no deeper insight into the nature of selenospecies was possible with SEC separation. The adoption of a previously published SAX-ICP-DRC-MS method17 was successful in speciating CSF samples with sufficient resolution and sensitivity. In total six seleno-compounds were found, from which SePP, TrxR, Se-HSA and selenate could be identified and quantified, whilst two more Se-peaks remained unidentified. Based on a) the SEC-ICP-DRC-MS result showing 95% of Se attributed to high molecular weight compounds (proteins) and only 5% to LMM compounds, and b) on quantification of these two unidentified seleno-species during SAX-ICP-DRC-MS, resulting in 27% or 20% of total Se (i.e. each > 5% from SEC), it is concluded that both unidentified Se-compounds are of HMM nature. Problems were seen for the chromatographic stability of SePP during SAX separation, leading to underestimation of this Se-species and overestimation of selenate. Surprisingly, GPx could not be found in the investigated CSF samples, which might be explained by instability of this compound during SAX separation, leading as well to overestimation of selenate.References
- P. D. Whanger, J Trace Elem Electrolytes Health Dis, 1992, 6, 209–221 Search PubMed.
- P. Morenoa, M. A. Quijano, A. M. Gutiérrez, M. C. Pérez-Conde and C. Cámara, Anal. Chim. Acta, 2004, 524, 315–327 CrossRef CAS.
- J. R. Arthur, F. Nicol and G. J. Beckett, Biol. Trace Elem. Res., 1992, 33, 37–42 CrossRef CAS.
- U. Schweizer, A. U. Bräuer, J. Köhrle, R. Nitsch and N. E. Savaskan, Brain Res. Rev., 2004, 45, 164–178 CrossRef CAS.
- J. K. Andersen, Nat. Med., 2004, 10/Suppl, S18–S25 CrossRef.
- P. Jenner, Ann. Neurol., 2003, 53(Suppl), S26–S36 CrossRef CAS.
- V. Aguilar, F. J. Jimenez-Jimenez, J. A. Molina, I. Meseguer, C. J. Mateos-Vega, M. J. Gonzales-Munoz, F. de Bustos, C. Gomez-Escalonilla, M. Orti-Pareia, M. Zurdo and M. C. Martinez-Para, J. Neural Transm., 1998, 105, 1245–1251 CrossRef.
- S. Fahn and G. Cohen, Ann. Neurol., 1992, 32, 802–812.
- M. Scharpf, U. Schweizer, T. Arzberger, W. Roggendorf, L. Schomburg and J. Köhrle, J. Neural Transm., 2007, 114, 877–884 CrossRef CAS.
- K. Renko, M. Werner, I. Renner-Müller, T. G. Cooper, C. H. Yeung, B. Hollenbach, M. Scharpf, J. Köhrle, L. Schomburg and U. Schweizer, Biochem. J., 2008, 409, 741–749 CrossRef CAS.
- http://www.neuropathologyweb.org/chapter14/chapter14CSF.html (28.07.2010).
- L. E. Walther, S. Streck, B. W. Walther, E. Treff, K. Winnefeld and E. Beleites, J Trace elements and electrolytes, 2000, 17/2, 87–91 Search PubMed.
- L. Gerhardsson, Th. Lundh, L. Minthon and E. Londos, Dementia Geriatr. Cognit. Disord., 2008, 25, 508–515 Search PubMed.
- B. Michalke, P. Grill and J. Berthele, J. Trace Elem. Med. Biol., 2009, 23, 243–250 CrossRef CAS.
- I. Meseguer, J. A. Molina, F. J. Jimenez-Jimenez, V. Aguilar, C. J. Mateos-Vega, M. J. Gonzales-Munoz, F. de Bustos, M. Orti-Pareia, M. Zurdo, A. Berbel, E. Barrios and M. C. Martinez-Para, J. Neural Transm., 1999, 106, 309–315 CrossRef CAS.
- K. Jitmanee, N. Teshima, T. Sakai and K. Grudpan, Talanta, 2007, 73/2, 352–357 CrossRef CAS.
- M. Xu, L. M. Yang and Q. Q. Wang, J. Anal. At. Spectrom., 2008, 23, 1545–1549 RSC.
- K. Shigeta, K. Sato and Furuta, J. Anal. At. Spectrom., 2007, 22, 911–916 RSC.
- V. Nischwitz, A. Berthele and B. Michalke, Anal. Chim. Acta, 2008, 627/2, 258–269 CrossRef CAS.
- O. Mikes, High performance liquid chromatography of biopolymers and biooligomers, Elsevier, 1988, Amsterdam, Oxford, New York, Tokyo Search PubMed.
- L. Flohe, B. Eisele and A. Wendel, Hoppe-Seyler′s Zeitschrifz für physiologische Chemie, 1971, 352/1, 151–158 Search PubMed.
- I. Nakamoto and K. Morimoto, Environ. Health Preventive Med., 2008, 9/2, 63–66 Search PubMed.
- M. Shuaguang, K. E. Hill, R. M. Caprioli and R. F. Burk, J. Biol. Chem., 2002, 277, 12749–12754 CrossRef CAS.
- B. Eberle and H. J. Haas, J. Trace Elem. Electrolytes Health Dis., 1993, 7/4, 217–221 Search PubMed.
- B. Hollenbach, N. G. Morgenthaler, J. Struck, C. Alonso, A. Bergmann, J. Köhrle and L. Schomburg, J. Trace Elem. Med. Biol., 2008, 22, 24–32 CrossRef CAS.
- K. Shigeta, K. Matsumura, Y. Suzuki, A. Shinohara and N. Furuta, Anal. Sci., 2008, 24, 1117–1122 CrossRef CAS.
- P. Jitaru, M. Prete, G. Cozzi, C. Turetta, W. Cairns, R. Seraglia, P. Tralfi and P. Cescon, J. Anal. At. Spectrom., 2008, 23, 402–406 RSC.
- J. Yan and J. N. Barrett, J. Neuroscience, 1998, 18/21, 8682–8691 CAS.
- E. S. Arnér and A. Holmgren, Eur. J. Biochem., 2000, 267/20, 6102–6109 CrossRef CAS.
- M. Kühbacher, J. Bartel, B. Hoppe, D. Alber, G. Bukalis, A. U. Bräuer, D. Behne and A. Kyriakopoulos, J. Neurochem., 2009, 110, 133–142 CrossRef.
- G. J. Pyne-Geithman, D. N. Caudell, P. Prakash and J. F. Clark, Neurol. Res., 2009, 31/2, 195–199 Search PubMed.
- R. A. Condell and A. L. Tappel, Arch. Biochem. Biophys., 1983, 223/2, 407–416 CrossRef CAS.
- W. B. Davidson, D. G. Kennedy, P. J. Hughes and W. J. Blanchflower, Vet. Res. Commun., 1990, 14/6, 441–446 CrossRef CAS.
- W. R. Zhang, P. K. Ku, E. R. Miller and D. E. Ullrey, Can J. Vet. Res., 1986, 50, 390–392 Search PubMed.
Footnote |
| † This article is part of a themed issue highlighting outstanding and emerging work in the area of speciation. |
| This journal is © The Royal Society of Chemistry 2011 |