First total synthesis of antrocamphin A and its analogs as anti-inflammatory and anti-platelet aggregation agents†
Chia-Lin
Lee
ah,
Chi-Huan
Huang‡
a,
Hui-Chun
Wang
a,
Da-Wei
Chuang
a,
Ming-Jung
Wu
b,
Sheng-Yang
Wang
c,
Tsong-Long
Hwang
d,
Chin-Chung
Wu
a,
Yeh-Long
Chen
e,
Fang-Rong
Chang
*af and
Yang-Chang
Wu
*agh
aGraduate Institute of Natural Products, Kaohsiung Medical University, Kaohsiung 807, Taiwan. E-mail: aaronfrc@kmu.edu.tw; Fax: +886 7 311 4773; Tel: +886 7 312 1101 ext. 2162
bDepartment of Chemistry, National Sun Yant-sen University, Kaohsiung 804, Taiwan
cDepartment of Forestry, National Chung-Hsing University, Taichung 402, Taiwan
dGraduate Institute of Natural Products, Chang Gung University, Tao-Yuan 333, Taiwan
eDepartment of Medicinal and Applied Chemistry, Kaohsiung Medical University, Kaohsiung 807, Taiwan
fDepartment of Marine Biotechnology and Resources, National Sun Yat-sen University, Kaohsiung 804, Taiwan
gGraduate Institute of Integrated Medicine, College of Chinese Medicine, China Medical University, Taichung 40402, Taiwan. E-mail: yachwu@mail.cmu.edu.tw; Fax: +886 4 220 60248; Tel: +886 4 220 57153
hNatural Medicinal Products Research Center, China Medical University Hospital, Taichung 40402, Taiwan
First published on 18th November 2010
Abstract
Naturally occurring antrocamphin A (1) is a potent anti-inflammatory compound from the edible fungus Antrodia camphorata (Taiwanofungus camphoratus), whose wild fruiting body is used as a valuable folk medicine in Taiwan. This study is the first total synthesis of antrocamphin A (1) and its analogs. Their inhibition ability on NO release, superoxide anion generation, elastase release and platelet aggregation are reported herein.
Introduction
The endemic fungus Antrodia camphorata (Taiwanofungus camphoratus), also known as Niu-Chang-Chih (Jang-Jy), is used as a folk medicine and a dietary supplement in Taiwan. Its wild fruiting body is very valuable.1–3 The chemical composition of this fungus can be classified into three categories: 1). Polysaccharides, 2). Triterpenoids and 3). Enynyl-benzenoids. The polysaccharides are regarded as immunomodulation agents, such as functional foods derived from other fungi, e.g., Ganoderma lucidum and Agaricus blazei.1Triterpenoids are considered to have anti-cancer and anti-inflammatory effects. However, the major chemical component, antrocamphin A (1) (Fig. 1), which belongs to the third class, is the key component for anti-inflammatory activity. Evidence is shown below through a complete bioassay-guided fractionation study.3,4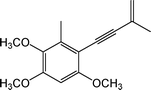 | ||
| Fig. 1 Antrocamphin A (1). | ||
Antrocamphin A (1), a potent anti-inflammatory compound naturally found in A. camphorata (T. camphoratus) and was first isolated by Chen et al. in 2007.4 Wang et al. demonstrated its mechanism in suppressing pro-inflammatory molecules (NO and PGE2), from being released via the down-regulation of iNOS and COX-2 expression through the NF-κB pathway.3 Previous studies indicate that compound 1 may serve as a promising lead drug for the treatment of various diseases that are induced by inflammation. Therefore, antrocamphin A was chosen to be a candidate of further investigation.
Currently, there are only two literature3,4 reports on the subject of antrocamphin A (1). To understand the diverse biological properties and the structure–activity relationship (SAR) of the lead compound 1, the first total synthesis of compound 1 and its analogs were achieved in the current investigation. All compounds were evaluated for anti-inflammatory and anti-platelet aggregation activities.
Results and discussion
The retrosynthetic analysis of compound 1 is illustrated in Scheme 1. The key step was the Sonogashira reaction. Compound 2 (2-iodo-3,5,6-trimethoxytoluene) was coupled with 2-methyl-1-buten-3-yne (3), which led to the target compound. Compound 2 was synthesized from 2,3,5-trimethoxytoluene (8), obtained from o-vanillin (5), in four steps described by Singh and co-workers.5 We prepared 2-hydroxy-3-methoxytoluene (4) by hydrogenating of o-vanillin (5) over 10% palladium-on-carbon. Compound 4 was then converted to quinone 6 by treatment with the oxidant, potassium nitrosodisulfonate (Fremy's salt). Quinone 6 was reduced to hydroquinone 7 using TiCl3 and then methylated with Me2SO4 in the presence of K2CO3 resulting in the desired compound 8. Compound (2), 2-iodo-3,5,6-trimethoxytoluene, a key synthon, was prepared viaiodination of 8 using I2 and CF3COOAg. The Sonogashira reaction was utilized to couple iodo 2 with 2-methyl-1-buten-3-yne (3), using the catalysts Pd(PPh3)4 and CuI. This key reaction led to the desired compound, antrocamphin A (1) (Scheme 2).6 The Sonogashira coupling reaction was also applied to afford a series of designed analogues (9–22) from commercially available iodobenzene products or further iodination benzenoid compounds (Scheme 3).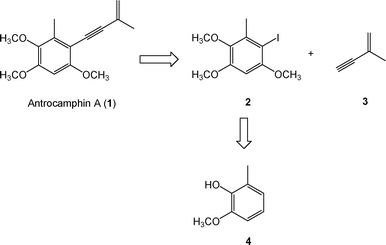 | ||
| Scheme 1 Retrosynthetic analysis of antrocamphin A. | ||
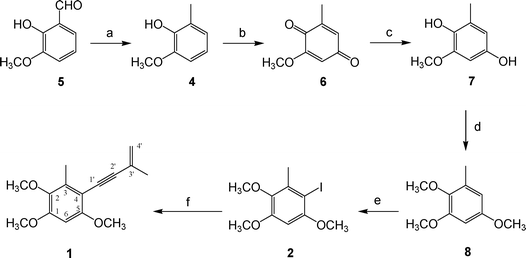 | ||
| Scheme 2 Synthetic scheme of antrocamphin A (1). Reagents and conditions: (a) 10% Pd/C, H2, rt, 75%; (b) KH2PO4, (KSO3)2NO, rt, 78%; (c) TiCl3, rt, 90%; (d) (CH3O)2SO2, K2CO3, acetone, reflux, 88%; (e) I2, CF3COOAg, CH2Cl2, 0 °C, 74%; (f) 2-methyl-1-buten-3-yne (3), Pd(PPh3)4, CuI, Et3N/THF, N2, rt, 10%. | ||
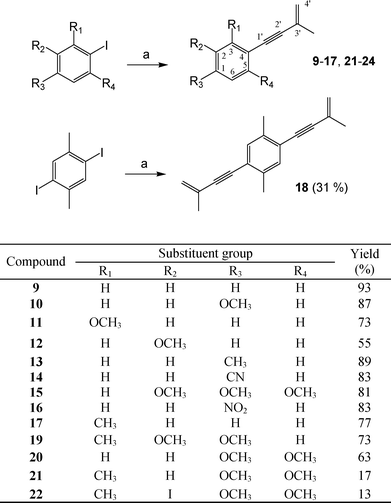 | ||
| Scheme 3 Synthesis of 9–22. Reagents and conditions: (a) 2-methyl-1-buten-3-yne (3), Pd(PPh3)4, CuI, Et3N/THF, N2, rt. | ||
Synthesized compounds 1 and 9–22 were screened using a nitric oxide (NO) inhibitory assay, where curcumin was used as the positive control. In this study, all analogs were tested on their cytotoxicity toward RAW 264.7 macrophages. Cell viability rates for each test compound showed > 90% at the dosage of 20 μg mL−1. All compounds were evaluated against lipopolysaccharide (LPS)-induced NO production in RAW 264.7 macrophage cell line at the dosage of 20 μg mL−1. As shown in Table 1, compounds 1, 20, 21 and 22 showed potent anti-inflammatory activity with NO inhibition rates of 98.1%, 97%, 77%, 84%, respectively.
| Compoundb | NO inhibition (%) | Cell viability (%) |
|---|---|---|
| a All compounds were administrated 1 h before inflammation induction by adding LPS (1 μg mL−1). b Dosage of test compound was 20 μg mL−1. c Positive control. | ||
| 1 | 98.10 | 99.71 |
| 9 | 17.23 | 101.55 |
| 10 | 33.56 | 101.98 |
| 11 | −4.43 | 90.44 |
| 12 | 27.27 | 105.80 |
| 13 | 23.11 | 100.95 |
| 14 | −0.55 | 91.77 |
| 15 | 24.98 | 101.91 |
| 16 | 15.16 | 94.97 |
| 17 | 6.23 | 99.73 |
| 18 | 16.61 | 92.23 |
| 19 | 28.51 | 99.07 |
| 20 | 97.00 | 117.08 |
| 21 | 77.00 | 114.00 |
| 22 | 84.00 | 102.00 |
| Curcumin c | 99.00 | 103.00 |
Compounds 1, 9, 15, 17, 20 and 21 were synthesized to study the SAR of the CH3 substitution at C-3 presented in the natural product 1; however, C3-CH3 did not influence the ability of NO inhibition. To evaluate the SAR of the OCH3 functions on the 3′-methyl-but-3′-en-1-ynyl-benzene skeleton, analogs 10–12, 15 and 20 were designed, which did not have the C3-CH3 substitution. Compounds 10–12 possess only one OCH3 group at C-1, C-3 and C-2, respectively. Moreover, 20 and 15 have two and three OCH3 groups at C-1 and C-5, and C-1, C-2 and C-5, respectively. Among the five compounds, we speculated that the position is more important than the number of OCH3 substitutions. A similar result was also observed in the test of compounds 19 and 21, which both have a CH3 group at C-3 and the same number of OCH3 substitutions in different positions. Furthermore, a comparison with the structures of 1 and 20–22 concluded that the simultaneous existence of OCH3 groups at C-1 and C-5 may be necessary for the potential ability of NO inhibition.
LPS-challenged RAW 264.7 macrophages are an in vitro model of murine cell lines for studying anti-inflammatory effects. To further explore the anti-inflammatory activities of these target compounds in human cell systems with different modes of action, all analogs were evaluated against superoxide anion generation and elastase release by human neutrophils in response toN-formyl-methionyl-leucyl-phenylalanine (FMLP)/cytochalasin B (CB). Antrocamphin A has been reported as a new anti-inflammatory drug lead against superoxide anion generation in FMLP-activated human neutrophils.4 Activated neutrophils produced high concentrations of the superoxide anion and elastase known to be involved in airway mucus hypersecretion, which is a neutrophilic inflammatory disease like asthma.7 Therefore, all analogues were tested for their ability to inhibit superoxide anion generation and elastase release. Diphenyleneiodonium (DPI) and phenylmethylsulfonyl fluoride (PMSF) were used as positive controls in this model assay, respectively.
Interestingly, the analogs showed a broad-spectrum of activity in this model (Table 2). Most of the compounds could inhibit both inflammatory mediators and showed more potent effects than the natural product 1. Compound 18 with a dienynyl functionality exhibited the best activity, which indicated the enynyl substitution played an important role for this model assay. On the whole, we found dimethoxy (19–22) and monomethoxy (10–12) derivatives were more favorable than trimethoxy and non-methoxy ones. Some of the compounds showed superior activities to the reference drug, PMSF, against elastase release by neutrophils.
| Compound | Superoxide | Elastase | ||
|---|---|---|---|---|
| IC50/μg mL−1a | or (Inh %) | IC50/μg mL−1a | or (Inh %) | |
| Per cent of inhibition (Inh%) at a 10-μg mL−1 concentration. Results are presented as mean ± S.E.M. (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001 compared with the control value.a Concentration necessary for 50% inhibition (IC50).b Diphenyleneiodonium (DPI) and phenylmethylsulfonyl fluoride (PMSF) were used as positive controls for superoxide anion generation and elastase release, respectively. | ||||
| 1 | (46.30 ± 7.24) | *** | (25.55 ± 3.99) | ** |
| 9 | 4.82 ± 0.41 | (37.22 ± 3.12) | *** | |
| 10 | 5.63 ± 0.71 | 4.14 ± 0.35 | ||
| 11 | 6.69 ± 0.84 | 7.94 ± 0.51 | ||
| 12 | 4.05 ± 0.41 | 3.31 ± 0.06 | ||
| 13 | 4.66 ± 0.41 | 44.28 ± 1.67 | *** | |
| 14 | (7.95 ± 6.06) | (17.89 ± 1.42) | *** | |
| 15 | 3.40 ± 1.49 | (26.74 ± 5.88) | * | |
| 16 | (−10.03 ± 4.84) | 22.78 ± 1.01 | *** | |
| 17 | (25.55 ± 6.70) | * | (35.55 ± 6.12) | ** |
| 18 | 0.45 ± 0.03 | 1.32 ± 0.67 | ||
| 19 | 1.96 ± 0.28 | 3.47 ± 0.23 | ||
| 20 | 4.69 ± 0.99 | 8.72 ± 1.15 | ||
| 21 | 1.60 ± 0.28 | 5.33 ± 1.21 | ||
| 22 | (12.13 ± 2.24) | ** | (81.36 ± 8.82) | |
| DPI b | 0.22 ± 0.13 | |||
| PMSF b | 22.80 ± 5.07 | |||
Furthermore, all synthesized derivatives were also subjected to an anti-platelet aggregation assay with collagen and thrombin as inducers; aspirin was used as a positive control. As shown in Table 3, compounds 9, 10, 11 and 19 showed potent inhibition against collagen-induced platelet aggregation with IC50 values of 3.36, 1.16, 7.55 and 7.22 μg mL−1, respectively; their activities were better than aspirin's activity (IC50 13.58 μg mL−1). Comparing the bioassay results, most of the compounds did not possess good inhibition toward the platelet aggregation induced by thrombin. No clear SAR can be concluded from the anti-platelet aggregation effect. Interestingly, compounds 1 and 20–22, which possess the powerful ability of NO inhibition, did not have anti-platelet aggregation activity.
Conclusions
In summary, we have achieved the first total synthesis of the active natural product antrocamphin A (1), in a total yield of 3.7% in six steps. Moreover, 14 analogs, including new compounds, 15, 16 and 18–22, were synthesized. Most of these compounds possess OMe groups in the benzene ring. We tried to synthesized derivatives with different substitutions around the ring, for example, 2-iodo-3,6-diacetoxy-5-methoxytoluene or 4-iodo-3,6-dihydroxy-5-methoxytoluene coupled with 2-methyl-1-buten-3-yne using different combinations of catalyst [Pd(PPh3)4 or PdCl2(PPh3)2], solvent (THF or DMF or ether) and base (Et3N). Unfortunately, this resulted in no reaction or reaction without any target compound. Accordingly, only the reagents with OMe groups, which were easily obtained, were chosen to evaluate the amount and position of OMe groups related to the activity in this investigation.Comparing anti-inflammatory activity, compounds 1, 20, 21 and 22 were identified to be potent NO inhibition agents; the dienynyl compound 18 was a new drug lead against superoxide anion generation and elastase release. Moreover, analog 10 is the most potential compound against platelet aggregation induced by collagen. Overall, our data demonstrate that enynyl-benzenoids may have a chance to be developed as safe and potential anti-inflammatory or a cardiovascular protecting agent in the future.
Acknowledgements
This work was supported by grants from National Science Council, Taiwan awarded to Y.-C. Wu and F.-R. Chang. This study is also supported in part by Taiwan Department of Health Clinical Trial and Research Center of Excellence (DOH99-TD-B-111-004) and Department of Health Cancer Research Center of Excellence (DOH99-TD-C-111-005).Notes and references
- M. C. Lu, S. L. Hwang, F. R. Chang, Y. H. Chen, C. S. Hung, C. L. Wang, Y. H. Chu, S. H. Pan and Y. C. Wu, Food Chem., 2009, 113, 1049 CrossRef CAS.
- M. C. Lu, Y. C. Du, J. J. Chuu, S. L. Hwang, P. C. Hsieh, F. R. Chang and Y. C. Wu, Arch. Toxicol., 2009, 83, 121 CrossRef CAS.
- Y. H. Hsieh, F. H. Chu, Y. S. Wang, S. C. Chien, S. T. Chang, J. F. Shaw, C. Y. Chen, W. W. Hsiao, Y. H. Kuo and S. Y. Wang, J. Agric. Food Chem., 2010, 58, 3153 CrossRef CAS.
- J. J. Chen, W. J. Lin, C. H. Liao and P. C. Shieh, J. Nat. Prod., 2007, 70, 989 CrossRef CAS.
- U. S. Singh, R. T. Scannell, H. An, B. J. Carter and S. M. Hecht, J. Am. Chem. Soc., 1995, 117, 12691 CrossRef CAS.
- M. L. Goddard and R. Tabacchi, Tetrahedron Lett., 2006, 47, 909 CrossRef CAS.
- T. L. Hwang, C. C. Wang, Y. H. Kuo, H. C. Huang, Y. C. Wu, L. M. Kuo and Y. H. Wu, Biochem. Pharmacol., 2010, 80, 1190 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and spectral data. See DOI: 10.1039/c0ob00616e |
| ‡ Equal contributions as first author. |
| This journal is © The Royal Society of Chemistry 2011 |