Per(poly)fluoroalkanesulfinamides assisted diastereoselective three-component inverse-electron-demand aza Diels–Alder reaction†
Peng
Li
,
Li-Juan
Liu
and
Jin-Tao
Liu
*
Key Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Lu, Shanghai 200032, China. E-mail: jtliu@mail.sioc.ac.cn; Fax: (+86) 21-6416-6128
First published on 17th November 2010
Abstract
A highly diastereoselective three-component inverse-electron-demand aza Diels–Alder reaction assisted by per(poly)fluoro-alkanesulfinamides is presented, providing a broad spectrum of highly functionalized piperidine derivatives with excellent endo/exo and facial diastereoselectivities. The electron-withdrawing perfluoroalkyl groups are crucial for the success of this reaction under mild conditions and facilitate monitoring the process and stereoselectivities of the reaction. The synthetic potential of these cycloadducts is also highlighted.
The asymmetric aza Diels–Alder reaction is among the most powerful and convergent strategies for the stereoselective construction of highly valuable piperidine derivatives.1,2 As a complementary alternative to the well established formal cycloaddition of dienes and imines, Boger and co-workers introduced inverse-eletron-demand aza Diels–Alder (IEDDA) reactions of N-sulfonyl-1-aza-1,3-butadienes and electron-rich alkenes,3 which is a very appealing strategy in view of the generally exhibited high regiospecificity and diastereoselectivity. In contrast to the formal cycloaddition, however, very few precedents have been disclosed on the asymmetric variants of these reactions, probably due to the low reactivity of 1-azadienes and high propensity of both azadienes and dienophiles to decompose in the presence of Lewis acids.4–7 Recently, Boger and co-workers developed an asymmetric IEDDA reaction of N-sulfonyl-1-aza-1,3-butadienes and enol ethers bearing chiral auxiliaries,5 and later asymmetric IEDDA reaction of 1-azadienes catalyzed by Lewis acids6 and organocatalysts7 were also reported, providing efficient protocols to access a wide variety of optically active compounds of the piperidine type. One drawback with these methods is, however, the removal of (hetero)arylsulfonyl groups, which requires harsh conditions.8
Up to now, the chiral auxiliary strategy is still an important technology in asymmetric synthesis from a practical point of view, because the separation of diastereomeric products prior to the cleavage of the chiral auxiliary provides enantiomerically pure products. Sulfinamides, as efficient chiral auxiliaries, have been widely used in the asymmetric synthesis of amines.9 One of their appealing advantages is the convenience to remove the sulfinyl residue from the products. However, their application in asymmetric IEDDA reaction of 1-azadienes met with significant challenges: (i) due to the lower electron-withdrawing ability of sulfinyl group compared to that of the sulfonyl moiety, the asymmetric IEDDA reaction of N-sulfinyl-1-aza-1,3-butadienes with electron-rich olefins was usually conducted under harsh thermal conditions (high temperature or high pressure). Accordingly, either the DA adducts could only exist as reaction intermediates and subsequent aromatization occurred automatically,10 or the facial selectivity of the reaction of specified substrates was rather low;11 (ii) the asymmetric three-component variant remains unknown. Therefore the development of novel chiral auxiliaries and new strategies to improve this reaction and extend the reaction scope is still of great significance.
Recently, we reported a diastereoselectivity-switchable and regio-specific hetero Diels–Alder reaction of N-sulfinyl per(poly)fluoroalkanesulfinamides with dienes, wherein the chiral induction potential of per(poly)fluoroalkanesulfinamides (PFSAs) was initially demonstrated.12 On this basis, we became interested in the synthetic potential of PFSAs as novel chiral auxiliaries. Herein we describe a highly diastereoselective three-component IEDDA reaction of α,β-unsaturated ketones, PFSAs and enol ethers. The success of this reaction relies on the strong electron-withdrawing ability of the per(poly)fluoroalkyl groups.
As a starting point, different per(poly)fluoroalkanesulfinamides 2a–d were prepared from per(poly)fluoroalkyl iodides according to the procedures previously developed by us.12 Then the three-component reaction of trifluoromethyl α,β-unsaturated ketone 1a, 2-chlorotetrafluoroethanesulfinamide (2a) and ethyl vinyl ether (3a) was investigated. After surveying the reaction conditions by varying the water scavenger, solvent, temperature and stoichiometry, we were pleased to find that the reaction occurred readily in CH2Cl2 at room temperature in the presence of 3.0 equiv. Ti(OiPr)4, affording the DA adduct 4a as a single diastereomer13 in 65% yield (Table 1, entry 1). Using the most popular tert-butanesulfinamide (2e) or sterically less demanding n-butanesulfinamide (2f) as chiral auxiliaries under otherwise identical conditions afforded no DA cycloadducts but the corresponding imine intermediates, undoubtedly owing to the low reactivity of the imine intermediates (Table 1, entries 5 and 6). Other PFSAs (2b–2d) were also employed in the three-component IEDDA reaction. All these reactions furnished the corresponding DA adducts with good to excellent endo/exo and facial diastereoselectivities (Table 1, entries 2–4). The reactivities of the sulfinimines produced in situ in the cycloaddition step were proportional to the electron-withdrawing abilities of per(poly)fluoroalkyl groups, which were observed in the following sequence: nC4F9 > CF3 ≈ ClF2CF2C > PhCF2. Among them, sulfinamide 2a provided the best facial selectivity.
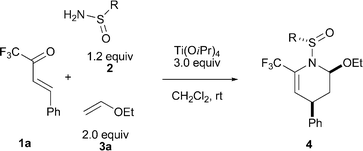
| entry | R (2) | 4 | yield (%)b |
endo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :exoc :exoc |
facial selectivityc | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Racemic sulfinamides were used. b Isolated yield of the major endo adduct after column chromatography. c Determined by 19F NMR and diagnostic 1H NMR. d Only the corresponding imine was isolated. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | ClF2CF2C (2a) | 4a | 65 | >35:1 |
21:1 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | CF3 (2b) | 4b | 63 | >50:1 |
10:1 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | PhCF2 (2c) | 4c | 42 | >50:1 |
18:1 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | nC4F9 (2d) | 4d | 59 | >40:1 |
19:1 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | tBu (2e) | — | —d | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | nBu (2f) | — | —d | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The scope of this three-component cycloaddition protocol was explored next. As shown in Table 2, a series of dienophiles were firstly evaluated in the three-component reaction with 1a and 2a under the optimized reaction conditions. In general, monosubstituted vinyl ethers with primary or secondary alkyl substituents at oxygen afforded the cycloadducts smoothly in good yields with good to excellent endo/exo and facial diastereoselectivities (Table 2, entries 1–3). Good diastereoselectivity was also observed when allenic ethyl ether (3e) was used as dienophile (Table 2, entry 4). However, in the case of cyclic vinyl ethers such as dihydrofuran (3f), no desired cycloadducts were obtained owing to the rapid decomposition of ethers in the presence of Lewis acid additive (Table 2, entry 5).
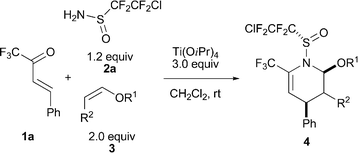
| entry | R1 | R2 | 4 | yield (%)b |
endo:exoc |
facial selectivityc | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Racemic sulfinamide 2a was used. b Isolated yield of the major endo adduct after column chromatography. c Determined by 19F NMR and diagnostic 1H NMR. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | iBu | H | 4e | 63 | >50:1 |
26:1 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | nBu | H | 4f | 67 | >80:1 |
25:1 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | Cy | H | 4g | 63 | 16:1 |
12:1 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | Et | ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 CH2 |
4h | 46 | 10:1 |
8:1 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | –C2H4– | — | — | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
As for the 1-azadiene counterpart, a variety of ketones 1a–1n were surveyed using isobutyl vinyl ether (3b) as the dienophiles under the above conditions (Table 3). Good yields and high levels of endo-selectivity and facial diastereoselectivity were achieved in most cases. Aryl substituents of varied electronic and steric nature at the β position (R3) were well tolerated (Table 3, entries 2–7), with the exception that the bulky naphthyl substituent remarkably lowered the facial selectivity of the corresponding DA adduct 4p (Table 3, entry 8). Decrease in diastereoselectivities was also observed in the asymmetric IEDDA reaction of ketones with aliphatic substituents at the β position (R3), and the yields were generally quite low with the emergence of byproducts formed by the direct DA reactions of α,β-unsaturated ketones and 3b (Table 3, entries 9–10). Substituents at the carbonyl carbon (R1) exhibited a pronounced effect on the endo-selectivity and facial diastereoselectivity. While the CO2iPr group greatly improved the endo-selectivity and facial diastereoselectivity of the reaction (Table 3, entry 12), the endo-selectivity and facial diastereoselectivity were relatively lower in the case of CF2Cl (Table 3, entry 11). Cinnamaldehyde (1m) without an electron-withdrawing group gave no DA adducts due to the low reactivity of the imine intermediate formed in the reaction (Table 3, entry 13). Although the endo/exo diastereoselectivity of the DA adduct of acrylonitrile (1n) was quite good, the facial diastereoselectivity was only 2.6:1, similar to that of the p-tolyl substituent counterpart (Table 3, entry 14).11 It is worth mentioning that the two endo products could be easily separated by column chromatography or recrystallization and obtained in pure form respectively.
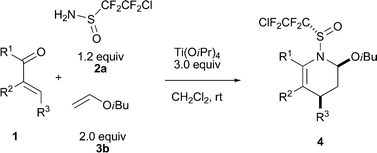
| entry | 1 | 4 | yield (%)b | endo:exoc | facial selectivityc | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
a Racemic sulfinamide 2a was used.
b Isolated yield of the major endo adduct after column chromatography.
c Determined by 19F NMR and diagnostic 1H NMR.
d The diastereoselectivities were further confirmed by GC-MS analysis of the crude products.
e Only the corresponding imine was isolated.
f Isolated yield of the minor endo product in parentheses.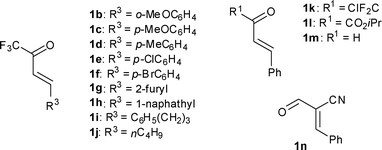
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | 1a | 4e | 63 | >50:1 |
26:1 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 1b | 4j | 65 | >60:1 |
21:1 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 1c | 4k | 59 | 43:1 |
21:1 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | 1d | 4l | 64 | 53:1 |
21:1 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | 1e | 4m | 67 | >40:1 |
50:1 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | 1f | 4n | 59 | 42:1 |
22:1 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | 1g | 4o | 46 | 38:1 |
21:1 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | 1h | 4p d | 60 | 26:1 |
14:1 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | 1i | 4q | 38 | 21:1 |
10:1 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | 1j | 4r | 32 | 21:1 |
17:1 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 | 1k | 4s | 47 | 27:1 |
17:1 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12 | 1l | 4t | 68 | >100:1 |
>100:1 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 13 | 1m | — | —e | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 14 | 1n | 4v | 57 (20)f | >100:1 |
2.6:1 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
To further demonstrate the utility of PFSAs as chiral auxiliaries in the asymmetric IEDDA reaction, sulfinamide (SS)-2a was successfully prepared in enantiopure form (>99% ee) by ammonia hydrolysis of 2-chlorotetrafluoroethanesulfinyl-oxazolidione (5)14 with LiHMDS (Scheme 1). The enantioselective three-component IEDDA reaction was then conducted with (SS)-2a and no racemization was observed with product (SS,2S,4R)-6 as shown by chiral HPLC analysis (>99.9% ee). The DA adduct (SS,2S,4R)-6 could be smoothly converted into a number of valuable compounds (Fig. 1). Limited attempts at direct desulfination of (SS, 2S,4R)-6 led only to decomposition.11 Upon heating in THF, however, disubstituted pyridine 7 was produced efficiently.10 The transformation of (SS,2S,4R)-6 into its 2-hydroxy derivative 8 was readily achieved in 58% yield with complete stereoselectivity15 by treatment with BCl3 in CH2Cl2, whereas the RO group was removed chemoselectively to give compound 9 by nucleophilic replacement with Et3SiH/BCl3 at ambient temperature. Deprotection of compound 9 was achieved conveniently with HCl/Et2O in MeOH. Without purification, intermediate imine 1016 was reduced with NaBH3CN directly to give a single diastereomer of trifluoromethylated piperidine 11,1a,17 a fluorinated common subunit found in compounds of varied pharmacological activities, in 71% total yield with 98% ee.
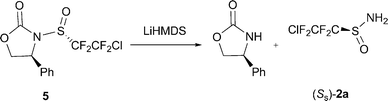 | ||
| Scheme 1 Preparation of sulfinamide (SS)-2a. | ||
In conclusion, we have developed a highly stereoselective three-component IEDDA reaction assisted by PFSAs, providing an efficient method for the synthesis of highly functionalized piperidine derivatives (especially trifluoromethylated ones18) with excellent endo/exo selectivities and facial diastereoselectivities. Initial experiments that highlight the synthetic potential of these piperidine derivatives and the convenient removal of the per(poly)fluoroalkanesulfinyl groups were also presented. It is noteworthy that the use of these PFSA auxiliaries made it possible to monitor the process and stereoselectivities of the reaction conveniently by 19F NMR spectroscopy. Moreover, the one-pot three-component reaction avoided the manipulation of the unstable imine intermediates, extending the scope of IEDDA reaction to specific substrates. Further studies on the applications of these PFSAs in the asymmetric synthesis are in progress in our laboratory.
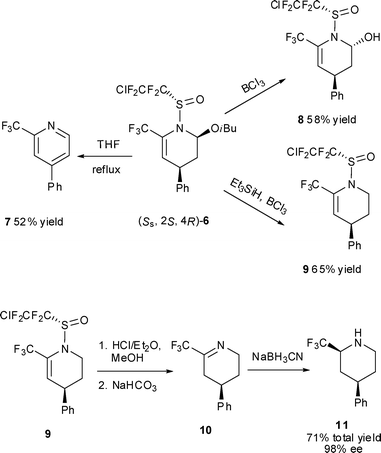 | ||
| Fig. 1 Synthetic potential of the cycloadducts. | ||
Acknowledgements
Financial support from the National Natural Science Foundation of China (No. 20872166) is gratefully acknowledged.Notes and references
- For reviews on the importance of piperidine derivatives, see: (a) P. D. Bailey, P. A. Millwood and P. D. Smith, Chem. Commun., 1998, 633 RSC; (b) M. Rubiralta, E. Giralt, A. Diez, Piperidine: Structure, Preparation, Reactivity, and Synthetic Applications of Piperidine and Its Derivatives, Elsevier, Amsterdam, 1991 Search PubMed.
- For recent reviews on stereoselective aza-Diels–Alder reactions, see: (a) For a review on catalytic asymmetric aza-Diels–Alder reactions, see: G. B. Rowland, E. B. Rowland, Q. Zhang and J. C. Antilla, Curr. Org. Chem., 2006, 10, 981 Search PubMed; (b) G. R. Heintzelman, I. R. Meigh, Y. Mahajan and S. M. Weinreb, Org. React., 2005, 65, 141 CAS; (c) S. Kobayashi, in Cycloaddition Reactions in Organic Synthesis (ed.: S. Kobayashi, K. A. Jørgensen), Wiley-VCH: Weinheim, 2002, Chap. 5, 187–209 Search PubMed.
- (a) D. L. Boger and A. M. Kasper, J. Am. Chem. Soc., 1989, 111, 1517 CrossRef CAS; (b) D. L. Boger, W. L. Corbett and J. M. Wiggins, J. Org. Chem., 1990, 55, 2999 CrossRef CAS; (c) D. L. Boger and T. T. Curran, J. Org. Chem., 1990, 55, 5439 CrossRef CAS; (d) D. L. Boger, W. L. Corbett, T. T. Curran and A. M. Kasper, J. Am. Chem. Soc., 1991, 113, 1713 CrossRef CAS; (e) D. L. Boger, K. C. Cassidy and S. Nakahara, J. Am. Chem. Soc., 1993, 115, 10733 CrossRef CAS; (f) J. Hong and D. L. Boger, J. Am. Chem. Soc., 1998, 120, 1218 CrossRef CAS; (g) M. J. Schnermann and D. L. Boger, J. Am. Chem. Soc., 2005, 127, 15704 CrossRef CAS; (h) for a review on the ADAR of 1-azadienes, see: M. Behforouz and M. Ahmadian, Tetrahedron, 2000, 56, 5259 Search PubMed.
- For selected examples on diastereoselective ADARs of 1-azadienes, see: (a) M. C. Aversa, A. Barattucci, M. C. Bilardo, P. Bonaccorsi and P. Giannetto, Synthesis, 2003, 2241 CrossRef CAS; (b) C. R. Berry and R. P. Hsung, Tetrahedron, 2004, 60, 7629 CrossRef CAS; (c) J. E. Tarver, Jr., K. M. Terranova and M. M. Joullié, Tetrahedron, 2004, 60, 10277 CrossRef.
- R. C. Clark, S. S. Pfeiffer and D. L. Boger, J. Am. Chem. Soc., 2006, 128, 2587 CrossRef CAS.
- (a) J. Esquivias, R. G. Arrayás and J. C. Carretero, J. Am. Chem. Soc., 2007, 129, 1480 CrossRef CAS; (b) J. Esquivias, I. Alonso, R. G. Arrayás and J. C. Carretero, Synthesis, 2009, 113 CAS.
- (a) M. He, J. R. Struble and J. W. Bode, J. Am. Chem. Soc., 2006, 128, 8418 CrossRef CAS; (b) B. Han, J.-L. Li, C. Ma, S.-J. Zhang and Y.-C. Chen, Angew. Chem., Int. Ed., 2008, 47, 9971 CrossRef CAS; (c) B. Han, Z.-Q. He, J.-L. Li, R. Li, K. Jiang, T.-Y. Liu and Y.-C. Chen, Angew. Chem., Int. Ed., 2009, 48, 5474 CrossRef CAS; (d) Z.-Q. He, B. Han, R. Li, L. Wu and Y.-C. Chen, Org. Biomol. Chem., 2010, 8, 755 RSC; (e) for a recent review, see: B. Groenendaal, E. Ruijter and R. V. A. Orru, Chem. Commun., 2008, 5474 Search PubMed.
- P. G. M. Wuts, T. W. Greene, Greene's Protective Groups in Organic Synthesis, 4th ed.; John Wiley & Sons: New Jersey, 2007 Search PubMed.
- For recent reviews, see for example: (a) F. A. Davis, P. Zhou and B.-C. Chen, Chem. Soc. Rev., 1998, 27, 13 RSC; (b) J. A. Ellman, T. D. Owens and T. P. Tang, Acc. Chem. Res., 2002, 35, 984 CrossRef CAS; (c) F. A. Davis, J. Org. Chem., 2006, 71, 8993 CrossRef CAS; (d) G.-Q. Lin, M.-H. Xu, Y.-W. Zhong and X.-W. Sun, Acc. Chem. Res., 2008, 41, 831 CrossRef CAS; (e) F. Ferreira, C. Botuha, F. Chemla and P. L. Alejandro, Chem. Soc. Rev., 2009, 38, 1162 RSC; (f) M. T. Robak, M. A. Herbage and J. A. Ellman, Chem. Rev., 2010, 110, 3600 CrossRef CAS.
- (a) F. Palacios, J. Vicario and D. Aparicio, Tetrahedron Lett., 2007, 48, 6747 CrossRef CAS; (b) Similar elimination–aromatization phenomena were reported as side reaction, see: S. Kobayashi, T. Furuya, T. Otani and T. Saito, Tetrahedron, 2008, 64, 9705 Search PubMed.
- L. F. Tietze and A. Schuffenhauer, Eur. J. Org. Chem., 1998, 1629 CrossRef CAS.
- X.-J. Wang and J.-T. Liu, Tetrahedron, 2005, 61, 6982 CrossRef CAS.
- The stereochemistry of 4 was unambiguously determined by X-ray analysis of 4c (CCDC 790386).
- V. D. Romanenko, C. Thoumazet, V. Lavallo, F. S. Tham and G. Bertrand, Chem. Commun., 2003, 1680 RSC.
- The 2,4-trans relative stereochemistry of 8 (CCDC 790387) was assigned by X-ray analysis.
- As a 14:1 mixture with its enamine isomer which was characterized by 19F NMR and crude 1H NMR.
- For the synthesis of racemic isomer of piperidine 10, see: A. Gheorghe, B. Quiclet-Sire, X. Vila and S. Z. Zard, Tetrahedron, 2007, 63, 7187 Search PubMed.
- For the asymmetric synthesis of trifluoromethylated aliphatic nitrogen heterocycles, see: (a) T. Okano, M. Fumoto, T. Kusukawa and M. Fujita, Org. Lett., 2002, 4, 1571 CrossRef CAS; (b) J. Jiang, R. J. DeVita, G. A. Doss, M. T. Goulet and M. J. Wyvratt, J. Am. Chem. Soc., 1999, 121, 593 CrossRef CAS; (c) P. Bravo, M. Crucianelli, A. Farina, S. V. Meille, A. Volonterio and M. Zanda, Eur. J. Org. Chem., 1998, 435 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectra and analytical data for the products. CCDC reference numbers 790386 (4c) and 790387 (8). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0ob00699h |
| This journal is © The Royal Society of Chemistry 2011 |