Total syntheses of subereamollines A and B†
James W.
Shearman
a,
Rebecca M.
Myers
a,
James D.
Brenton
b and
Steven V.
Ley
*a
aDepartment of Chemistry, Lensfield Road, University of Cambridge, Cambridge, United Kingdom CB2 1EW. E-mail: svl1000@cam.ac.uk; Fax: +44 1223 3363968
bCancer Research UK Cambridge Research Institute, Li Ka Shing Centre, Robinson Way, Cambridge, United Kingdom CB2 0RE
First published on 15th October 2010
Abstract
The first total syntheses of (+)- and (−)-subereamollines A and B are reported. The enantiomeric forms of the natural products were obtained by preparative chiral HPLC separation of the corresponding racemates.
Marine sponges of the order Verongida are a rich source of bromotyrosine-derived natural products. Many of these compounds are biologically active and exhibit a range of activities including antiviral,1 antimicrobial,2 antifungal,3 Na+/K+ ATPase inhibition,4 anti-HIV,5 HDAC inhibition,6 antifouling,7 histamine H3 antagonism,8 mycothiol S-conjugate amidase inhibition,9 and isoprenylcysteine carboxy methyltransferase (Icmt) inhibition.10 Anticancer activity has also been reported, for example, the spirocyclohexadienylisoxazolines purealidin P and Q are cytotoxic against murine lymphoma K1210 (IC50 2.8 and 0.95 μg mL−1 respectively) and human epidermal carcinoma KB (nasopharynx) (IC50 7.6 and 1.2 μg mL−1 respectively) cell lines.11,12 Other structurally related compounds have also exhibited cytotoxic properties.13
When the extracts of the Red Sea sponge Suberea mollis were reinvestigated recently, two new bromotyrosine-derived secondary metabolites containing the spirocyclohexadienylisoxazoline moiety were identified; subereamollines A (1) and B (2) (Scheme 1).14 Small quantities (2–5 mg) were isolated and 1 was found to have modest antimicrobial activity against Staphylococcus aureus, however, their cytotoxic activity was not reported. As part of our ongoing investigation into the anticancer properties of bromotyrosine-derived natural products,15 the subereamollines were attractive synthetic targets.
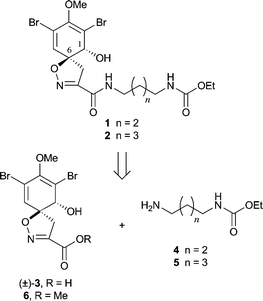 | ||
| Scheme 1 Retrosynthetic analysis of subereamollines A (1) and B (2). | ||
Both 1 and 2 contain a 2,4-dibromo-1-hydroxy-3-methoxyspirocyclohexadienylisoxazole moiety attached to a carbamate containing side chain via an amide linkage. Although not explicitly stated in the isolation paper,14 the absolute stereochemistries at C(1) and C(6) were determined to be (R) and (S) based on the optical rotations of 1 {[α]D +156.5 (c 0.55, MeOH)} and 2 {[α]D +22.9 (c 6.25, CH2Cl2)}. These data correlated with that reported for (+)-aerothionin {[α]D +210 (c 1.7, MeOH)} whose absolute configuration was determined by X-ray crystallography and circular dichroism (CD) spectroscopy.16
We envisaged that 1 and 2 could be synthesised by coupling spiro acid (±)-3 with either amine 4 or 5 (Scheme 1). Interestingly, 3 has been isolated from the marine sponge Pseudoceratina sp. as an optically pure isomer with its absolute configuration defined as 1S,6R by CD analysis.17 However, the only reported synthesis of (±)-3 is as an intermediate during the synthesis of purealin.18 The corresponding methyl ester 619–22 has been synthesised in an enantioenriched form (74% ee) via an asymmetric oxidative spirocyclisation which required a multi-step synthesis of a chiral auxiliary.23 An alternative approach led to enantiopure 6 by resolution of (±)-6 by derivatisation with (−)-camphanic chloride.24
Our approach to the naturally occurring enantiomers of 1 and 2 involved chiral HPLC separation of the corresponding racemates as this would require no additional synthetic operations and would also deliver enantiopure material required for biological screening.
Aldehyde 8 was prepared in 92% yield over two steps via bromination of 2-hydroxy-4-methoxybenzaldehyde (7) with N-bromosuccinimide and subsequent benzyl protection of the phenolic oxygen (Scheme 2). Conversion of 8 into the corresponding azlactone 9 was achieved by heating the aldehyde with N-acetylglycine and sodium acetate in the presence of acetic anhydride (Erlenmeyer conditions).25 Saponification of the crude azlactone 9 with barium hydroxide and subsequent condensation with O-benzylhydroxylamine furnished the carboxylic acid 10 in 49% yield. This yield did not improve when 9 recrystallised from acetone was used. Furthermore, oxime 11 was discovered to be a major by-product (22% yield), which may have arisen from reversion of 9 to its parent aldehyde under the basic conditions and subsequent condensation with the O-benzylhydroxylamine. Azlactones are known to hydrolyse efficiently under acidic conditions (e.g. 3 N HCl, reflux, 24 h)26 to give α-keto acid derivatives which can then be converted to the corresponding oxime acids.27 However, when these conditions were applied to 9 this unexpectedly resulted in the loss of the phenolic benzyl protecting group and spontaneous lactonisation to produce coumarin 12 (in 68% yield) as the only observable product.28
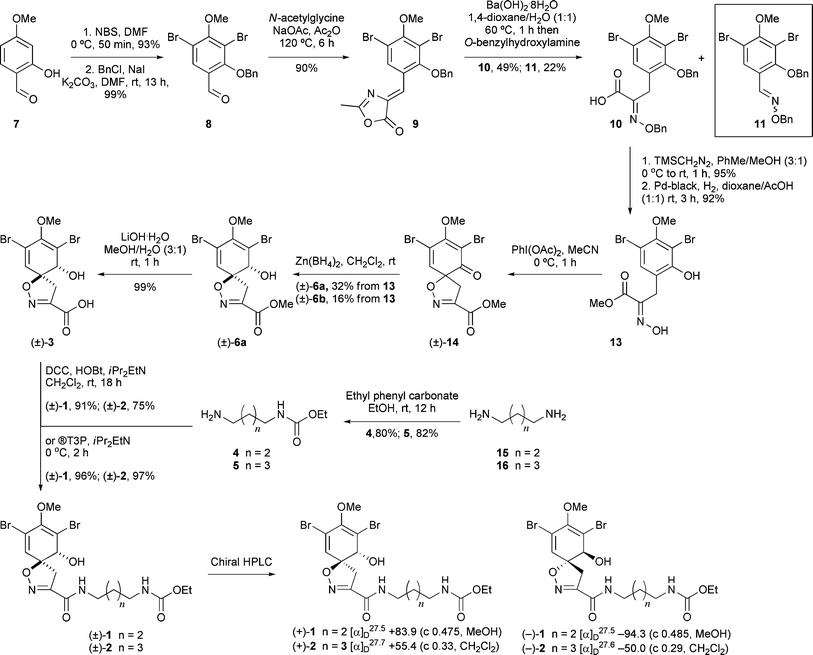 | ||
| Scheme 2 Synthesis route to 1 and 2. | ||
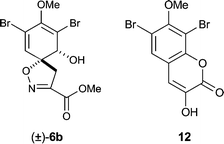
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr over the undesired cis-isomer (±)-6b. X-Ray crystallographic analysis confirmed the relative stereochemistry of each isomer.30 The methyl ester was then hydrolysed with lithium hydroxide to furnish (±)-3 in an overall yield of 11% from aldehyde 7.
:1 dr over the undesired cis-isomer (±)-6b. X-Ray crystallographic analysis confirmed the relative stereochemistry of each isomer.30 The methyl ester was then hydrolysed with lithium hydroxide to furnish (±)-3 in an overall yield of 11% from aldehyde 7.
Putrescine (15) and cadaverine (16) were converted to the amine coupling partners 4 and 5 in one step via dropwise addition of ethyl phenyl carbonate31 to minimize bis-carbamylation. Spiro acid (±)-3 was then coupled to amine 4 or 5 in the presence of N,N′-dicyclohexylcarbodiimide (DCC) and hydroxybenzotriazole (HOBt) to afford the natural products subereamolline A (1) and B (2) respectively in 91% and 75% yield as a racemic mixture. Upon reinvestigation of this coupling step, it was found that the use of the coupling agent propylphosphonic anhydride (®T3P) furnished 1 and 2 in 96% and 97% yields respectively.
The 1H and 13C NMR spectra of synthetic 1 and 2 matched those of the natural materials.32 We have also established that the originally reported 13C chemical shift assignments for C-2 (δC 122.9) and C-4 (δC 114.3) should be switched on the basis of COLOC NMR experiments reported for related spirocyclised products.33 This is also supported by the HMBC correlations in the natural products.34
The enantiomeric forms of (±)-1 and (±)-2 were separated using preparative chiral HPLC (CHIRALPAK AD-H column and 10% iPr2OH in hexane at a flow rate of 5 mL min−1) (Scheme 2). The optical rotations of (+)-1 (t = 40.3 min) and (+)-2 (t = 48.3 min) agreed with those observed for natural 1 and 2. To confirm the absolute stereochemistries at C(1) and C(6), (+)-6 (t = 30.9 min) and (−)-6 (t = 32.9 min) were obtained from chiral HPLC separation of (±)-6 (Scheme 3).35 Hydrolysis of (+)-6 and (−)-6 under the basic conditions furnished acids (+)-3 and (−)-3 that were then coupled with amines 4 and 5 respectively. HPLC analysis of the products showed them to be (+)-1 and (−)-2 respectively (see Electronic Supplementary Information), thus confirming the original stereochemical assignments at C(1) and C(6).
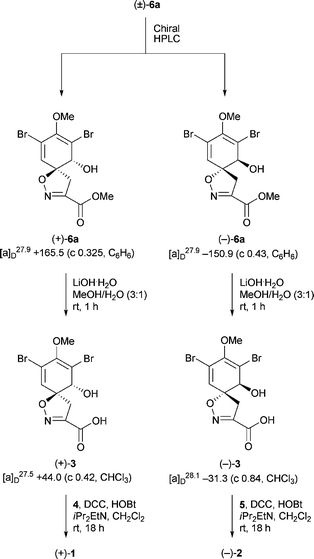 | ||
| Scheme 3 | ||
MTS assays36 were performed to evaluate the cytotoxic activity of compounds (±)-1, (±)-2, (+)-1, (+)-2, 12 and 13 against an ovarian cancer (SK-OV-3) cell line. Following 48 h treatment, none of the compounds exhibited cytotoxic activities up to a concentration of 100 μM.
In conclusion, the first total syntheses of the bromotyrosine-derived natural products subereamolline A (1) and B (2) have been reported. The enantiomeric forms of 1 and 2 were obtained by chiral HPLC separation and, in addition, the stereochemistries at C(1) and C(6) in the naturally occurring enantiomers (+)-1 and (+)-2 were confirmed to be R and S respectively.
Acknowledgements
We acknowledge the Cambridge Cancer Research UK PhD Training Programme in Medicinal Chemistry for funding (J.W.S.). The authors thank Dr John E. Davies of the X-ray facility of the Department of Chemistry for collecting the crystallographic data and Dr Richard M. Turner for help with preparative chiral HPLC.Notes and references
- S. P. Gunasekera and S. S. Cross, J. Nat. Prod., 1992, 55, 509 CrossRef CAS.
- J. Kobayashi, M. Tsuda, K. Agemi, H. Shigemori, M. Ishibashi, T. Sasaki and Y. Mikami, Tetrahedron, 1991, 47, 6617 CrossRef CAS.
- J.-H. Jang, R. W. M. van Soest, N. Fusetani and S. Matsunaga, J. Org. Chem., 2007, 72, 1211 CrossRef CAS.
- H. Nakamura, H. Wu and J. Kobayashi, Tetrahedron Lett., 1985, 26, 4517 CrossRef CAS.
- S. A. Ross, J. D. Weete, R. F. Schinazi, S. S. Wirtz, P. Tharnish, P. J. Scheuer and M. T. Hamann, J. Nat. Prod., 2000, 63, 501 CrossRef CAS.
- M. W. B. McCulloch, G. S. Coombs, N. Banerjee, T. S. Bugni, K. M. Cannon, M. K. Harper, C. A. Veltri, D. M. Virshup and C. M. Ireland, Bioorg. Med. Chem., 2009, 17, 2189 CrossRef CAS.
- I. Thironet, D. Daloze and J. C. Braekman, Nat. Prod. Lett., 1998, 12, 209 CrossRef.
- R. A. K. Mierzwa, M. A. Conover, S. Tozzi, M. S. Puar, M. Patel and S. J. Covan, J. Nat. Prod., 1994, 57, 175 CrossRef CAS.
- G. M. Nicholas, G. L. Newton, R. C. Fahey and C. A. Bewley, Org. Lett., 2001, 3, 1543 CrossRef CAS.
- M. S. Buchanan, A. R. Carroll, G. A. Fechner, A. Boyle, M. Simpson, R. Addepalli, V. M. Avery, J. N. A. Hooper, T. Cheung, H. Chen and R. J. Quinn, J. Nat. Prod., 2008, 71, 1066 CrossRef CAS.
- T. Fujiwara, J.-H. Hwang, A. Kanamoto, H. Nagai, M. Takagi and S.-Y. Kauzo, J. Antibiot (Tokyo), 2009, 62, 393 CrossRef CAS; P. B. Shinde, Y. M. Lee, H. T. Dang, J. Hong, C.-O. Lee and J. H. Jung, Bioorg. Med. Chem. Lett., 2008, 18, 6414 CrossRef CAS.
- J. Kobayashi, K. Honma, T. Sasaki and M. Tsuda, Chem. Pharm. Bull., 1995, 43, 403 CAS.
- J. Peng, J. Li and M. T. Hamann, Alkaloids, 2005, 61, 59 Search PubMed.
- M. I. Abou-Shoer, L. A. Shaala, D. T. A. Youssef, J. M. Badr and A.-A. M. Habib, J. Nat. Prod., 2008, 71, 1464 CrossRef CAS.
- J. W. Shearman, R. M. Myers, T. M. Beale, J. D. Brenton and S. V. Ley, Tetrahedron Lett., 2010, 51, 4812 CrossRef CAS.
- J. A. McMillan, I. C. Paul, Y. M. Goo and K. L. Rinehart Jr, Tetrahedron Lett., 1981, 22, 39 CrossRef.
- A. Aiello, E. Fattorusso, M. Menna and M. Pansini, Biochem. Syst. Ecol., 1995, 23, 377 CrossRef CAS.
- G. Zhu, F. Yang, R. Balachandran, P. Höök, R. B. Vallee, D. P. Curran and B. W. Day, J. Med. Chem., 2006, 49, 2063 CrossRef CAS.
- S. Nishiyama and S. Yamamura, Tetrahedron Lett., 1983, 24, 3351 CrossRef CAS.
- T. Ogamino, Y. Ishikawa and S. Nishiyama, Heterocycles, 2003, 61, 73 CrossRef CAS.
- O. Hoshino, M. Masatoshi and Y. Kohei, Tetrahedron, 1996, 52, 14713 CrossRef.
- T. R. Boehlow, J. J. Harburn and C. D. Spilling, J. Org. Chem., 2001, 66, 3111 CrossRef CAS.
- M. Murakata, M. Tamura and O. Hoshino, J. Org. Chem., 1997, 62, 4428 CrossRef CAS.
- T. Ogamino, R. Obata and S. Nishiyama, Tetrahedron Lett., 2006, 47, 727 CrossRef CAS.
- E. Erlenmeyer, Justus Liebigs Ann. Chem., 1893, 275, 1 CrossRef.
- B. M. Chanda and R. S. Sulake, Tetrahedron Lett., 2005, 46, 6461 CrossRef CAS; P. Busca, F. Paradisi, E. Moynihan, A. R. Maguire and P. C. Engel, Org. Biomol. Chem., 2004, 2, 2684 RSC.
- N. Kotoku, H. Tsujita, A. Hiramatsu, C. Mori, N. Koizumi and M. Kobayashi, Tetrahedron, 2005, 61, 7211 CrossRef CAS.
- Crystal data for 12: C10H6Br2O4, M = 349.97, orthorhombic, space group Pna2(1), a = 8.9710(2) Å, b = 10.6189(2) Å, c = 22.6185(6) Å, V = 2154.69(8) Å3, Z = 8, Dc = 2.158 Mg m−3, T = 180(2) K. The X-ray structure of coumarin 12 has been previously reported. J. J. Harburn, N. P. Rath and C. D. Spilling, J. Org. Chem., 2005, 70, 6398 Search PubMed.
- Prepared using literature procedure. W. J. Gensler, F. A. Johnson and D. B. Sloan, J. Am. Chem. Soc., 1960, 82, 6074 Search PubMed.
-
Crystal data for (±)-6a: C11H11Br2NO5, M = 397.03, triclinic, space group P
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 6.5189(1) Å, b = 10.7194(2) Å, c = 11.0482(2) Å, V = 684.99(2) Å3, Z = 2, Dc = 1.925 Mg m−3, T = 180(2) K. CCDC 787666. Crystal data for (±)-6b: C11H11Br2NO5, M = 397.03, monoclinic, space group P-2(1)/c, a = 10.9772(1) Å, b = 25.2605(3) Å, c = 10.1567(2) Å, V = 2725.4(3) Å3, Z = 8, Dc = 1.935 Mg m−3, T = 180(2) K. CCDC 787667.
, a = 6.5189(1) Å, b = 10.7194(2) Å, c = 11.0482(2) Å, V = 684.99(2) Å3, Z = 2, Dc = 1.925 Mg m−3, T = 180(2) K. CCDC 787666. Crystal data for (±)-6b: C11H11Br2NO5, M = 397.03, monoclinic, space group P-2(1)/c, a = 10.9772(1) Å, b = 25.2605(3) Å, c = 10.1567(2) Å, V = 2725.4(3) Å3, Z = 8, Dc = 1.935 Mg m−3, T = 180(2) K. CCDC 787667. - Prepared using literature procedure. G. Bartoli, M. Bosco, A. Carlone, M. Locatelli, E. Marcantoni, P. Melchiorre, P. Palazzi and L. Sambri, L., Eur. J. Org. Chem., 2006, 4429 Search PubMed.
- See Supplementary Information for 1H and 13C NMR spectra of natural and synthetic 1 and 2.
- P. Ciminiello, V. Costantino, E. Fattorusso, S. Magno, A. Mangoni and M. Pansini, J. Nat. Prod., 1994, 57, 705 CrossRef CAS.
- In the HMBC spectra of the natural and synthetic samples of 1 and 2, the quaternary carbon (δC 114.3) couples strongly to the protons H-1 (δH 4.07 and 4.06 respectively) and H-5 (δH 6.42 and 6.40 respectively). However, the quaternary carbon (δC 122.9) only couples to H-5. Therefore the original assignments for C-2 (δC122.9) and C-4 (δC 114.3) proposed by Habib and co-workers should be switched.
- Both (+)-6a (t = 30.9 min) and (−)-6a (t = 32.9 min) were converted to their corresponding camphanate esters and their NMR spectral data matched that reported by Nishiyama and co-workers (see Ref. 24).
- The (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (MTS) assay gives a colourimetric measure of the number of viable cells present in vitro.
Footnote |
| † Electronic supplementary information (ESI) available: experimental procedures for the preparation of compounds 1–6 and 8–14 and full characterisation of these compounds. 1H and 13C NMR spectra of 1 and 2. CCDC reference numbers 787666 and 787667. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0ob00636j |
| This journal is © The Royal Society of Chemistry 2011 |