The formation of core cross-linked star polymer and nanogel assemblies facilitated by the formation of dynamic covalent imine bonds†
Alexander W.
Jackson
,
Christopher
Stakes
and
David A.
Fulton
*
Chemical Nanoscience Laboratory, School of Chemistry, Newcastle University, Bedson Building, Newcastle upon Tyne, UK. E-mail: d.a.fulton@ncl.ac.uk; Fax: +44 (0)191 222 6929; Tel: +44 (0)191 222 7065
First published on 30th August 2011
Abstract
Reversible addition-fragmentation chain transfer (RAFT) polymerization has been utilised to prepare diblock copolymer chains possessing ‘inert’ blocks of polystyrene and ‘reactive’ blocks displaying aldehyde or amino functional groups. These polymer chains were shown to cross-link through the formation of imine bonds in organic solvents to form kinetically stable polymer assemblies possessing core-cross-linked star (CCS) polymers architectures which display a size-dependency upon the concentration at which the cross-linking reactions are performed. Evidence was found for the formation of a 2-armed species as a side-product in the formation of CCS polymers. RAFT polymerization was utilised to prepare a series of methacrylate copolymers possessing only ‘reactive’ blocks displaying a significantly lower density of aldehyde and amino functional groups than the diblock copolymers utilized in the formation of CCS polymers. These copolymers were shown to cross-link through the formation of imine bonds in organic solvents to form polymer assemblies possessing nanogel architectures. The lower density of functional groups was required to promote the formation of discrete nanogel assemblies. This cross-linking also displayed a size-dependency upon the concentration at which the cross-linking reactions are performed. Macroscopic gelation of these polymer chains was observed at higher concentrations of the copolymer chains. Both the CCS polymers and nanogels underwent structural reconfiguration to linear polymer chains through component exchange facilitated by trans-imination with a small molecule amine. We speculate that the formation of both the CCS polymers and nanogels is under kinetic control on account of the lack of reversibility of imine bonds in organic solvents.
Introduction
Recent years have seen the utilization of dynamic bonds, whether they be non-covalent supramolecular interactions or so-called dynamic covalent bonds, to endow polymeric systems with the abilities to adapt their structures or compositions in response to an external stimuli.1 When dynamic bonds are incorporated into polymeric systems, their reversible nature enables these systems to modify their architectures by reshuffling, incorporating or releasing their components, in effect providing a mechanism for polymer-based assemblies to reconfigure their molecular structures and therefore their functional or material properties. This design trend2 has lead to the emergence of impressive new materials possessing thermoresponsive,3 chemoresponsive,4 mechanoresponsive,5 photoresponsive,6 electroresponsive7 or self-healing properties.8Polymer nanoparticles represent an important class of polymer materials, finding application in numerous technological applications such as electronics,9a,b coatings,9c and drug delivery.9d,e At Newcastle we have begun a research programme aimed at incorporating supramolecular interactions into polymer nanoparticles. In this work, we describe our efforts to incorporate dynamic covalent linkages into two classes of interesting and potentially useful polymer architectures which can be considered as nanoparticles, namely, core cross-linked star (CCS) polymers10 and nanogels.11 CCS Polymers are a class of nano-sized polymer assembly whose structural features are defined by a core consisting of a network of cross-linked polymer chains surrounded by a corona of polymeric arms. Much of the interest in CCS polymers is as a consequence of their architectures, possessing cores which can be utilized as carriers for small molecules such as drugs or fragrances, and coronal chains which help to solubilise and shield the cargo from its external environment. CCS Polymers also possess solubilities and viscosities similar to linear polymers of relatively low molecular weight, additional properties which make them potentially very useful in a diverse range of fields, most notably drug delivery12 as well as imaging13 and catalysis.14 Nanogels can be considered as CCS polymers without coronal arms, consisting only of a spherical network of cross-linked polymer chains. This class of polymer architecture has been extensively investigated as drug delivery carriers on account of their high stabilities and loading capacities.15
Our synthetic approach (Fig. 1) towards CCS polymers and nanogels is to cross-link preformed linear polymer chains through supramolecular interactions. Of particular interest to us is the dynamic covalent bond,16 as this supramolecular interaction is reversible but with the virtue that the strength of the covalent bond ensures product assemblies possess chemical robustness. Furthermore, dynamic covalent reactions are usually performed with the help of a suitable catalyst to aid kinetics and so the option exists to halt these reversible processes and kinetically “fix” the products simply by quenching the catalyst, an option which is not available within supramolecular systems where the equilibrium cannot be easily “turned-off”. Furthermore, recent advances in living radical polymerization methods17 allow the preparation of polymers possessing controlled positioning of functional groups, composition and molecular weights, facilitating the preparation of exquisitely precise building blocks for use in the construction of these nano-sized polymer architectures.
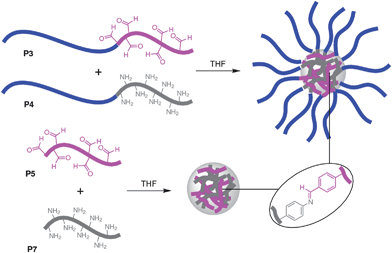 | ||
| Fig. 1 The formation of core cross-linked star polymer and nanogel nanoparticles facilitated by the cross-linking of polymer chains through the formation of imine bonds. | ||
The group of Otasuka and Takahara have demonstrated how dynamic covalent radical cross-over reactions can be used to prepare nanogel18 and CCS polymers19 possessing the abilities to reconfigure their structures at elevated temperatures. The group of Thayumanavan have utilized20 the reversible properties of the disulfide linkage to prepare nanogels which show potential in the fields of drug delivery. In this article we show that dynamic covalent imine bonds can be used to cross-link linear polymer chains into CCS polymer and nanogel nanoparticles. Imine condensation reactions are a particularly appealing dynamic covalent reaction as it is possible to tune the stability of the imine bond by altering stereoelectronic characteristics of the reaction partners, in particular the carbonyl-derived part.21 We demonstrate that the size of the nanoparticle can be determined simply by controlling the concentration at which the cross-linking is formed, and that the density of the cross-linking functional groups within the polymer building blocks has an important effect in promoting the formation of discrete nanoparticles. We also demonstrate the ability of these nanoparticles to undergo component exchange and reconfigure their structural properties from large cross-linked species to linear polymer chains.
Experimental
Materials and characterization
All chemicals were purchased from Sigma-Aldrich or Alfa Aesar and were used as received without further purification. 1H and 13C-NMR spectra were recorded on a Bruker Avance 300 spectrometer at 300 and 75 MHz, respectively, with the residual solvent signal as an internal standard. FTIR spectroscopy was performed on a Varian 800 FTIR instrument (Varian Inc.). High-resolution mass spectrometry was performed on a Waters LCT premier mass spectrometer (Waters Inc.). Gel permeation chromatography (GPC) was conducted on a Varian ProStar instrument (Varian Inc.) equipped with a pair of PL gel 5 μm Mixed D 300 × 7.5 mm columns with guard column (Polymer Laboratories Inc.) in series, a Varian 325 UV-vis dual wavelength detector (254 nm), a Dawn Heleos II multi-angle laser light scattering detector (Wyatt Technology Corp.) and a Viscotek 3580 differential RI detector. Near monodisperse methyl methacrylate or styrene standards (Agilent Technologies) were used for calibration. Data collection was performed with Galaxie software (Varian Inc.) and chromatograms analyzed with the Cirrus software (Varian Inc.) and Astra software (Wyatt Technology Corp.). MALDI-TOF analysis was performed on a Bruker Microflex (Bruker Daltonics). Samples for analysis were prepared by mixing solutions of dithranol (20 mg mL−1) and AgTFA (10 mg mL−1) with solutions of the polymer sample (10 mg mL−1) in volumes of 100 μL: 40μL: 20μL respectively. 0.6 μL of solution was spotted onto the MALDI plate, allowed to dry and then analyszed. Calibration was performed against near monodisperse polystyrene standards. Data collection was performed with Flexcontrol and analysis of spectra was performed using Flexanalysis software.4-Vinylbenzaldehyde22 (1)
A solution of 4-vinylbenzyl chloride (20.0 g, 130.2 mmol) and sodium acetate (12.9 g, 157.1 mmol) in (CH3)2SO (60 mL) was heated at 45 °C whilst stirring under N2 for 24 h. The reaction mixture was transferred to a separating funnel and H2O (100 mL) added. The product was extracted into EtOAc (3 × 100 mL). The organic extracts were combined and dried over MgSO4, filtered and evaporated to dryness to afford crude 4-vinylbenzylacetate as a pale yellow oil (24.8 g, 99%), which was dried under high vacuum. 20% NaOH(aq) (50 mL) was then added to a stirred solution of crude 4-vinylbenzylacetate (24.8 g, 140.8 mmol) in EtOH (50 mL) and the reaction mixture was heated at reflux for 4 h, allowed to cool to room temperature and then transferred into a separating funnel with EtOAc (100 mL). The aqueous layer was washed with EtOAc (2 × 100 mL), and the organic extracts were combined and dried over MgSO4, filtered and evaporated to dryness to afford crude 4-vinylbenzylalcohol as a brown oil (26.4 g, 100%) which was dried under high vacuum. A solution of (COCl)2 (8.6 g, 67.4 mmol) in CH2Cl2 (50 mL) was cooled to −78 °C whilst stirring under N2. A solution of (CH3)2SO (10.7 g, 134.8 mmol) in CH2Cl2 (50 mL) was added dropwise over 15 min, and a solution of crude 4-vinylbenzylalcohol (8.2 g, 61.3 mmol) in CH2Cl2 (50 mL) was then added dropwise over 30 min. Et3N (19.8 g, 196.0 mmol) was added to the reaction mixture and the solution was allowed to reach room temperature. The reaction mixture was left to stir under N2 for 16 h, then transferred into a separating funnel with HCl(aq) (1 M, 100 mL) and H2O (100 mL). The organic layer was collected and dried over MgSO4, filtered and evaporated to dryness to afford crude brown oil (8.1 g, 100%). The crude product was purified by vacuum distillation to afford 4-vinylbenzaldehyde as a colourless oil (4.2 g, 52%). 1H NMR (CDCl3): δ 5.44 (d, 1H, J = 10.8 Hz), 5.92 (d, 1H, J = 17.4 Hz), 6.76 (dd, 1H, J = 10.8 Hz, J = 17.4 Hz), 7.55 (d, 2H, J = 8.4 Hz), 7.84 (d, 2H, J = 8.4 Hz), 9.99 (s, 1H).N-Boc-4-aminostyrene23 (2)
A solution of 4-aminostyrene (4.5 g, 37.6 mmol) and di-tert-butyl dicarbonate (9.9 g, 45.2 mmol) in THF (100 mL) was heated at reflux under N2 for 16 h. After evaporation to dryness crude material was transferred to a separating funnel in H2O (50 mL) and the product was extracted with CH2Cl2 (3 × 50 mL). The organic extracts were combined and dried over Na2SO4, filtered, and evaporated to dryness to obtain a crude solid which was purified by column chromatography [SiO2, Hexane–EtOAc (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)] to afford N-Boc-4-aminostyrene as a white solid (6.7 g, 80%). 1H NMR (CDCl3): δ 1.52 (s, 9H), 5.15 (d, 1H, J = 9.9 Hz), 5.65 (d, 1H, J = 16.5 Hz), 6.53 (s, 1H), 6.65 (dd, 1H, J = 9.9 Hz, J = 16.5 Hz), 7.33 (s, 4H).
:1)] to afford N-Boc-4-aminostyrene as a white solid (6.7 g, 80%). 1H NMR (CDCl3): δ 1.52 (s, 9H), 5.15 (d, 1H, J = 9.9 Hz), 5.65 (d, 1H, J = 16.5 Hz), 6.53 (s, 1H), 6.65 (dd, 1H, J = 9.9 Hz, J = 16.5 Hz), 7.33 (s, 4H).
p-(2-Hydroxyethoxy)benzaldehyde (3)
Potassium carbonate (17.0 g, 0.123 mol) was suspended in a solution of 4-hydroxybenzaldehyde (5.0 g, 0.041 mol), 2-bromoethanol (5.1 g, 0.041 mol) and DMF (100 mL) under reflux conditions for 24 h. After filtration and evaporation to dryness, crude material was transferred into a separating funnel in CH2Cl2 (100 mL). The organic layer was washed with NaCl(aq) (2 × 100 mL), dried over MgSO4, filtered and evaporated to dryness to obtain a crude solid which was purified by column chromatography [SiO2, Hexane–EtOAc (1:1)] to afford p-(2-hydroxyethoxy)-benzaldehyde (1) as a colourless oil (4.2 g, 62%). 1H NMR (CDCl3): δ 3.10 (br s, 1H), 3.95 (t, 2H, J = 4.8 Hz), 4.11 (t, 2H, J = 4.8 Hz), 6.95 (d, 2H, J = 8.7 Hz), 7.75 (d, 2H, J = 8.7 Hz), 9.79 (s, 1H). 13C NMR (CDCl3): δ 61.4, 71.0, 115.2, 130.4, 132.4, 164.2, 191.5. FT-IR (wavenumber, cm−1): 3403 (O–H), 2934 (C–H), 2747 (C–H), 1676 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1509 (CC), 1427 (CC). HRMS+ C9H11O3: Theoretical: 167.0708. Actual: 167.0709.
O), 1509 (CC), 1427 (CC). HRMS+ C9H11O3: Theoretical: 167.0708. Actual: 167.0709.
p-(2-Methacryloxyethoxy)benzaldehyde (M1)
A solution of p-(2-hydroxyethoxy)benzaldehyde (3) (3.8 g, 0.023 mol) and Et3N (2.8 g, 0.027 mol) in CH2Cl2 (75 mL) was cooled to 0 °C. To this mixture a solution of methacryloyl chloride (2.7 g, 0.026 mol) in CH2Cl2 (25 mL) was added dropwise over 30 min whilst stirring under an atmosphere of N2. The reaction mixture was then allowed to reach room temperature and was left to stir for 2 h before transferring to a separating funnel with H2O (100 mL). The organic layer was washed with NaCl(aq) (2 × 100 mL), dried over MgSO4, filtered and evaporated to dryness to obtain a crude oil which was purified by column chromatography [SiO2, Hexane–EtOAc (4:1)] to afford p-(2-methacrylateethoxy)-benzaldehyde (M1) as a white solid (2.8 g, 52%). 1H NMR (CDCl3): δ 1.92 (s, 3H), 4.29 (t, 2H, J = 5. 1 Hz), 4.51 (t, 2H, J = 5.1 Hz), 5.57 (s, 1H), 6.12 (s, 1H), 7.00 (d, 2H, J = 8.7 Hz), 7.81 (d, 2H, J = 8.7 Hz), 9.86 (s, 1H). 13C NMR (CDCl3): δ 18.7, 63.0, 66.6, 115.3, 126.7, 130.7, 132.3, 136.2, 163.9, 167.5, 191.1. FT-IR (wavenumber, cm−1): 2981 (C–H), 2735 (C–H), 1694 (CO), 1509 (CC), 1449 (CC). HRMS+ C13H15O4: Theoretical: 235.0970. Actual: 235.0972.
N-Boc-4-aminophenol (4)
A solution of 4-aminophenol (12.6 g, 0.115 mol) and di-tert-butyl dicarbonate (30.3 g, 0.139 mol) in THF (100 mL) was stirred at 40 °C for 1.5 h. The reaction mixture was allowed to cool to room temperature and then filtered and evaporated to dryness to afford a pale yellow solid which was re-dissolved in EtOAc (150 mL) and transferred to a separating funnel with H2O (150 mL). The organic layer was dried over Na2SO4, filtered and evaporated to dryness to obtain a crude solid which was purified by recrystallization (EtOAc/CH2Cl2) to afford N-Boc-4-aminophenol (4) as a white solid (18.2 g, 76%). 1H NMR (CD3CN): δ 1.46 (s, 9H), 6.73 (d, 2H, J = 8.7 Hz), 6.79 (s, 1H), 7.20 (d, 2H, J = 8.7 Hz), 7.29 (s, 1H). 13C NMR (CD3CN): δ 27.3, 78.8, 114.9, 117.0, 120.5, 131.0, 152.3. FT-IR (wavenumber, cm−1): 3400 (O–H), 3352 (N–H), 2981 (C–H), 1695 (CO), 1505 (CC), 1436 (CC). HRMS+ C11H15O3NNa: Theoretical: 232.0950. Actual: 232.0949.
p-(2-Hydroxyethoxy)-N-Boc-aminobenzene (5)
Potassium carbonate (11.2 g, 0.081 mol) was added to a solution of N-Boc-4-aminophenol (4) (5.7 g, 0.027 mol) and 2-bromoethanol (10.2 g, 0.082 mol) in DMF (100 mL) and the resulting suspension heated under reflux conditions for 24 h. After filtration and evaporation to dryness, the crude material was dissolved in EtOAc (100 mL) and transferred into a separating funnel. The organic layer was washed with NaCl(aq) (2 × 100 mL), dried over Na2SO4, filtered and evaporated to dryness to obtain a crude oil which was purified by column chromatography [SiO2, Hexane–EtOAc (3:1)] to afford p-(2-hydroxyethoxy)-N-Boc-aminobenzene (5) as a white solid (4.2 g, 62%). 1H NMR (CDCl3): δ 1.50 (s, 9H), 2.61 (s, 1H), 3.91 (t, 2H, J = 4.8 Hz), 4.01 (t, 2H, J = 4.8 Hz), 6.66 (s, 1H), 6.82 (d, 2H, J = 9 Hz), 7.25 (d, 2H, J = 9 Hz). 13C NMR (CD3CN): δ 28.7, 61.8, 64.8, 70.3, 80,6, 115.6, 121.2, 132.5, 153.6. FT-IR (wavenumber, cm−1): 3400 (O–H), 3360 (N–H), 2982 (C–H), 2932 (C–H), 1695 (CO), 1518 (CC), 1459 (CC). HRMS+ C13H19O4NNa: Theoretical: 276.1212. Actual: 276.1211.
p-(2-Methacryloxyethoxy)-N-Boc-aminobenzene (M2)
A solution of p-(2-hydroxyethoxy)-N-Boc-aminobenzene (5) (2.9 g, 0.011 mol) and Et3N (1.4 g, 0.014 mol) in CH2Cl2 (75 mL) was cooled to 0 °C. A solution of methacryloyl chloride (1.3 g, 0.012 mol) in CH2Cl2 (25 mL) was added dropwise over 30 min whilst stirring under N2 and the reaction mixture was allowed to reach room temperature and then stirred for 2 h before transferring to a separating funnel with H2O (100 mL). The organic layer was washed with NaCl(aq) (2 × 100 mL), dried over Na2SO4, filtered and evaporated to dryness to obtain a crude oil which was purified by column chromatography [SiO2, Hexane–EtOAc (9:1)] to afford p-(2-methacryloxyethoxy)-N-Boc-aminobenzene (M2) as a white solid (2.3 g, 65%). 1H NMR (CDCl3): δ 1.51 (s, 9H), 1.95 (s, 3H), 4.18 (t, 2H, J = 5.1 Hz), 4.48 (t, 2H, J = 5.1 Hz), 5.59 (m, 1H), 6.15 (s, 1H), 6.56 (s, 1H), 6.85 (d, 2H, J = 9 Hz), 7.28 (d, 2H, 9 Hz). 13C NMR (CDCl3): 18.4, 27.8, 63.5, 67.1, 80.6, 115.8, 121.1, 126.0, 132.6, 136.6, 153.5, 155.2, 167.6. FT-IR (wavenumber, cm−1): 3331 (N–H), 2981 (C–H), 1694 (CO), 1525 (CC), 1463 (CC). HRMS+ C17H23O5NNa: Theoretical: 344.1474. Actual: 344.1488.
Aldehyde-functionalized copolymer (P1)
S-1-Dodecyl-S′-(α,α-dimethyl-α′′-acetic acid)trithiocarbonate (DDMAT) (1 eq, 57.5 mg, 0.158 mmol) and AIBN (0.2 eq, 5.2 mg, 31.6 μmol) were added to a small schlenk tube. Styrene (65 eq, 1.07 g, 10.3 mmol) and 4-vinylbenzaldehyde (65 eq, 1.37 g, 10.3 mmol) was then added followed by dioxane (2.5 mL). The reaction mixture was degassed five times, and then the vessel was backfilled with N2, purged with N2, and allowed to warm to room temperature. The reaction mixture was then placed in an oil bath at 70 °C, and the polymerization was quenched after 21 h and solvent was removed on the rotary evaporator. The resulting yellow oil was dissolved in a minimal amount of THF and added dropwise to a large excess of ice-cold hexane. The resulting polymer precipitate was then isolated by filtration and dried under high vacuum. Polymer P1 was obtained as a pale yellow solid (1.77 g, 91%). 1H NMR (CDCl3): δ 0.84 (br, SC11H22CH3, of the chain terminus), 1.42 (br, CHCH2, polymer backbone), 1.61 (br, CHCH2, polymer backbone), 3.29 (br, SCH2C11H23, of the chain terminus), 6.51 (br, Ar, polymer backbone), 7.03 (br, Ar, polymer backbone), 7.50 (br, Ar, polymer backbone), 9.89 (br, CHO, polymer backbone). The composition of P1 can be determined simply by comparing the integration of the aldehyde protons with the total integration of the aromatic protons. The monomer composition was determined to be 1:1 styrene:4-vinylbenzaldehyde.
N-Boc-protected amine-functionalized copolymer (P2a)
S-1-Dodecyl-S′-(α,α-dimethyl-α′′-acetic acid)trithiocarbonate (DDMAT) (1 eq, 52.1 mg, 0.143 mmol) and AIBN (0.2 eq, 4.7 mg, 28.6 μmol) were added to a small schlenk tube. Styrene (75 eq, 1.12 g, 10.7 mmol) and N-Boc-4-aminostyrene (75 eq, 2.35 g, 10.7 mmol) were then added followed by dioxane (3 mL). The reaction mixture was degassed five times and then the vessel was backfilled with N2, purged with N2, and allowed to warm to room temperature. The reaction mixture was then placed in an oil bath at 70 °C, and the polymerization was quenched after 22 h and solvent was removed on the rotary evaporator. The resulting yellow oil was dissolved in a minimal amount of THF and added dropwise to a large excess of ice-cold hexane. The resulting polymer precipitate was then isolated by filtration and dried under high vacuum. Polymer P2a was obtained as a pale yellow solid (1.18 g, 62%). 1H NMR (CDCl3): δ 0.89 (br, SC11H22CH3, of the chain terminus), 1.38 (br, CHCH2, polymer backbone), 1.55 (br, C(CH3)3), 1.76 (br, CHCH2, polymer backbone), 3.29 (br, SCH2C11H23, of the chain terminus), 6.53 (br, Ar, polymer backbone), 7.05 (br, Ar, polymer backbone). The composition of P2a can be determined simply by comparing the integration of the Boc protons at δ = 1.55 ppm with the total integration of the aromatic protons. The monomer composition was determined to be 1:1 styrene:N-Boc-4-aminosytene.
Amine-functionalized copolymer (P2b)
TFA (1 mL) was added to a stirred solution of P2a (0.1 g, 7.53 μM) in CH2Cl2 (1 mL) at room temperature. After 1 h the reaction mixture was evaporated to dryness to afford a crude yellow oil which was dissolved in minimal amount of THF and added dropwise to a large excess of ice-cold hexane. The resulting polymer precipitate was then isolated by filtration and dried under high vacuum. Polymer P2b was obtained as a pale yellow solid (0.09 g, 87%). 1H NMR (THF-d8): δ 0.85 (br, SC11H22CH3, of the chain terminus), 1.45 (br, CHCH2, polymer backbone), 1.79 (br, CHCH2, polymer backbone), 3.29 (br, SCH2C11H23, of the chain terminus), 6.56 (br, Ar, polymer backbone), 7.02 (br, Ar, polymer backbone), 7.44 (br, Ar, polymer backbone).Aldehyde-functionalized diblock copolymer (P3)
P1 (1 eq, 1.02 g, 85 μmol) and AIBN (0.2 eq, 2.8 mg, 17 μmol) were added to a small schlenk tube. Styrene (300 eq, 2.66 g, 25.5 mmol) was then added followed by dioxane (3 mL) and the reaction mixture was degassed five times. The vessel was backfilled with N2, purged with N2, and allowed to warm to room temperature. The reaction mixture was then placed in an oil bath at 70 °C, and the polymerization was quenched after 24 h and solvent was removed on the rotary evaporator. The resulting yellow oil was dissolved in a minimal amount of THF and added dropwise to a large excess of ice-cold methanol. The resulting polymer precipitate was then isolated by filtration and dried under high vacuum. Polymer P3 was obtained as a pale yellow solid (3.03 g, 96%). 1H NMR (CDCl3): δ 0.89 (br, SC11H22CH3, of the chain terminus), 1.43 (br, CHCH2, polymer backbone), 1.86 (br, CHCH2, polymer backbone), 6.57 (br, Ar, polymer backbone), 7.05 (br, Ar, polymer backbone), 7.52 (br, Ar, polymer backbone), 9.89 (br, CHO, polymer backbone).Amine-functionalized diblock copolymer (P4)
P2a (1 eq, 1.05 g, 79 μmol) and AIBN (0.2 eq, 2.6 mg, 15.8 μmol) were added to a small schlenk tube. Styrene (400 eq, 3.29 g, 31.6 mmol) was then added followed by dioxane (4 mL) and the reaction mixture was degassed five times. The vessel was backfilled with N2, purged with N2, and allowed to warm to room temperature. The reaction mixture was then placed in an oil bath at 70 °C, and the polymerization was quenched after 25.5 h and solvent was removed on the rotary evaporator. The resulting yellow oil was dissolved in a minimal amount of THF and added dropwise to a large excess of ice-cold methanol. The resulting polymer precipitate was then isolated by filtration and dried under high vacuum. The pale yellow solid obtained was dissolved in CH2Cl2 (3 mL) and TFA (3 mL) was added, solution was left to stir at room temperature. The reaction mixture was evaporated to dryness after 1 h, and the resulting crude yellow oil was dissolved in minimal amount of THF and added dropwise to a large excess of ice-cold hexane. The polymer precipitate was then isolated by filtration and dried under high vacuum. Polymer P4 was obtained as a pale yellow solid (1.11 g, 47%). 1H NMR (CDCl3): δ 0.89 (br, SC11H22CH3, of the chain terminus), 1.44 (br, CHCH2, polymer backbone), 1.87 (br, CHCH2, polymer backbone), 3.29 (br, SCH2C11H23, of the chain terminus), 6.59 (br, Ar, polymer backbone), 7.05 (br, Ar, polymer backbone).Aldehyde-functionalized copolymer (P5)
4-(4-Cyanopentanoic acid)dithiobenzoate (CPADB) (1 eq, 17.33 mg, 0.062 mmol) and AIBN (0.15 eq, 1.53 mg, 9.3 μmol) were added to a small schlenk tube. Methyl methacrylate (224 eq, 1.39 g, 13.89 mmol) and p-(2-methacryloxyethoxy)benzaldehyde (M1, 28 eq, 407 mg, 1.74 mmol) were then added followed by benzene (2.5 mL). The reaction mixture was degassed five times, and then the vessel was backfilled with N2, purged with N2, and allowed to warm to room temperature. The reaction mixture was then placed in an oil bath at 70 °C, and the polymerization was quenched after 24 h. The reaction mixture was dissolved in a minimal amount of THF and added dropwise to a large excess of hexane. The resulting polymer precipitate was then isolated by filtration and the precipitation was repeated before drying under high vacuum. Polymer P5 was obtained as a pale red solid (0.84 g). 1H NMR (CDCl3): 0.84 (br, C(CH3), polymer backbone), 1.80 (br, CH2, polymer backbone), 3.59 (br, OCH3), 4.31 (br, CH2CH2), 7.05 (br, Ar), 7.86 (br, Ar), 9.91 (br, CHO). The composition of P5 can be determined by comparing the integration of the aldehyde protons of M1 with the integration of the OCH3 protons of methyl methacrylate (MMA). The monomer composition was determined to be 8:1 MMA:M1 (identical to the feed ratio).
N-Boc-protected amine-functionalized copolymer (P6)
4-(4-Cyanopentanoic acid)dithiobenzoate (CPADB) (1 eq, 19.7 mg, 0.071 mmol) and AIBN (0.15 eq, 1.73 mg, 10.5 μmol) were added to a small schlenk tube. Methyl methacrylate (224 eq, 1.58 g, 15.8 mmol) and p-(2-methacryloxyethoxy)-N-Boc-aminobenzene (M2) (28 eq, 0.634 mg, 1.97 mmol) were then added followed by benzene (2.5 mL). The reaction mixture was degassed five times, and then the reaction vessel was backfilled with N2, purged with N2, and allowed to warm to room temperature then placed in an oil bath at 70 °C. The polymerization was quenched after 24 h and the reaction mixture was dissolved in a minimal amount of THF and added dropwise to a large excess of hexane. The resulting polymer precipitate was then isolated by filtration and the precipitation was repeated before drying under high vacuum. Polymer P6 was obtained as a pale red solid (1.36 g). 1H NMR (CDCl3): 0.85 (br, C(CH3), polymer backbone), 1.50 (br, C(CH3)3), 1.80 (br, CH2, polymer backbone), 3.59 (br, OCH3), 4.20 (br, CH2CH2), 6.69 (br, NH), 6.85 (br, Ar), 7.30 (br, Ar). The composition of P6 can be determined by comparing the integration of the aromatic protons of M2 with the integration of the OCH3 protons of methyl methacrylate (MMA). The monomer composition was determined to be 8:1 MMA:M2 (identical to the feed ratio).
Amine-functionalized copolymer (P7)
P6 (0.63 g) was dissolved in CH2Cl2 (5 mL) and TFA (3 mL) and the reaction mixture stirred for 2 h. The solvent and excess TFA were removed on the rotary evaporator and the oil obtained was dissolved in a minimal amount of THF and added dropwise to a large excess of hexane. The resulting polymer precipitate was then isolated by filtration and dried under high vacuum. Polymer P7 was obtained as a pale red solid (0.59 g). 1H NMR (THF-d8): 0.86 (br, C(CH3), polymer backbone), 1.83 (br, CH2, polymer backbone), 3.56 (br, OCH3), 4.21 (br, CH2CH2), 6.90 (br, Ar), 7.05 (br, Ar).Procedure for macroscopic gelation between P1 and P2b
Solutions of P1 (0.5 and 5 wt %) in THF with TFA (10 eq) were prepared and allowed to stir at room temperature for 10 min to allow complete dissolution of the polymer sample. Solutions of P2b (0.5 and 5 wt %) were prepared in THF with TFA (10 eq) under identical conditions. Equal volumes of these corresponding solutions were mixed and gelation was observed in both cases after 15 min and 2h respectively.Procedure for the preparation of CCS polymers (1–6)
Solutions of the aldehyde functional diblock copolymer component P3 (0.5–5 wt %) were prepared in THF with TFA (10 eq). Solutions of the amine functional diblock copolymer component P4 (0.5–5 wt %) were also prepared under identical conditions. Equal volumes of corresponding solutions of the same concentration were combined with rapid stirring and left to equilibrate at room temperature for 16 h before GPC–MALLS analysis. GPC analysis of all crude reaction mixtures reveals no evidence for any unreacted reactive polymers, indicating that the cross-linking reaction proceeds with 100% conversion.Procedure for the disassembly of CCS polymers
A sample of CCS polymers (2 mL, containing 2 wt % of polymer building blocks) was isolated. To this solution a large excess of propylamine (10 eq per aldehyde functional group) was added. TFA (0.1 eq with respect to propylamine) was then added and the reaction mixture was left to stir overnight at room temperature before GPC analysis.Procedure for the preparation of nanogels (7–12)
Equimolar solutions of P5 and P7 in THF with TFA (10 eq) were combined under rapid stirring to afford a range of solutions with a total polymer concentration of 0.1–2 wt %. Solutions were left to equilibrate at room temperature for 24 h before GPC–MALLS analysis. GPC Analysis of all crude reaction mixtures reveals no evidence for any unreacted polymers, indicating that the cross-linking reaction proceeds with 100% conversion.Procedure for the disassembly of nanogels
An excess of ethanolamine (10 eq per aldehyde functional group) was added to a solution of nanogels formed at 1 wt % of total copolymer building blocks. TFA (0.1 eq with respect to ethanolamine) was then added and the solution left to equilibrate at room temperature for 24 h before GPC analysis.Procedure for macroscopic gelation between P5 and P7
Equimolar solutions of P5 and P7 in THF with TFA (10 eq) were mixed to afford a range of solutions with a total polymer concentration of 3, 4 and 5 wt %. Solutions were left to equilibrate at room temperature for 24 h.Results and discussion
Polystyrene-based core cross-linked star (CCS) polymers
The first part of our work focused on investigating the potential to form CCS polymers by cross-linking styrenic block co-polymer chains P3 and P4 through imine bond formation. The polymers were prepared by RAFT polymerization, which was utilized throughout this study on account of its versatility and experimental simplicity. The RAFT chain transfer agent S-1-dodecyl-S′-(α,α-dimethyl-α′′-acetic acid)trithiocarbonate24 (DDMAT) was used (Scheme 1) to mediate the copolymerization of a 1:1 mixture of styrene and 4-vinylbenzaldehyde at 70 °C in dioxane to afford P1, which after purification was used as a macroinitiator and extended via RAFT polymerization with styrene at 70 °C in dioxane to afford P3 which features a ‘reactive block’ featuring approximately 50 aldehyde functions and an ‘inert’ block of polystyrene. Comparison of the GPC trace of the chain-extended polymer P3 with the macroinitiator P1 (Fig. 2a) indicates complete and successful chain extension. The copolymer P2a was prepared via RAFT polymerization of a 1:1 mixture of N-Boc-4-aminostyrene and styrene at 70 °C in dioxane, which was then used as a macroinitiator and extended via RAFT polymerization with styrene at 70 °C in dioxane to afford, after the removal of the protecting groups, P4. Again, comparison of the GPC trace of the chain-extended polymer P4 with the macroinitiator P2a (Fig. 2b) indicates complete and successful chain extension.25 A sample of the copolymer P2a was also subjected to acidic conditions to remove the protecting groups to yield P2b. All polymers P1–P4 were characterized by 1H NMR spectroscopy and gel permeation chromatography (GPC) (Table 1). The PDIs were found to be <1.40, indicating all polymerizations proceeded with a high level of control. The values of hydrodynamic radius (Rh) are consistent with linear polymer chains and indicate that there is no aggregation between the polymers when dissolved in THF.
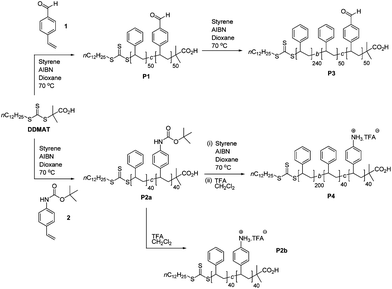 | ||
| Scheme 1 Synthetic route to aldehyde- and amine-functionalized styrenic diblock copolymers P1–P4. | ||
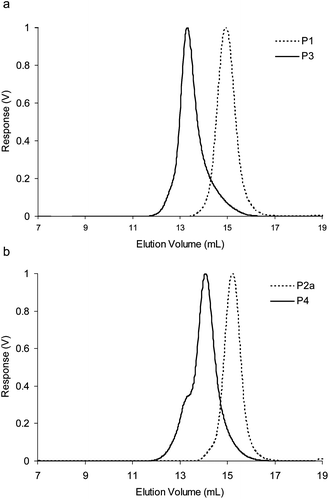 | ||
| Fig. 2 (a) Differential refractive index (dRI) GPC traces (THF 1.0 mL min−1) of P1 and resulting diblock copolymer P3 after chain extension, (b) dRI GPC traces (THF 1.0 mL min−1) of P2a and resulting diblock copolymer P4 after chain extension. | ||
| Polymer | M n (g mol−1) | M n (g mol−1) | M w (g mol−1) | PDIb (Mw/Mn) | R h (nm) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a As determined by 1H NMR spectroscopy. b As determined by gel permeation chromatography in THF (1.0 mL min−1) calibrated against polystyrene standards. c As determined by online dynamic LS measurements. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P1 | 12,200 | 9,200 | 10,750 | 1.17 | 2.5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P2a | 13,300 | 7,350 | 8,300 | 1.13 | 2.4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P2b | 13,800 | 8,000 | 9,000 | 1.13 | 2.6 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P3 | 37,200 | 32,000 | 43,300 | 1.35 | 4.5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P4 | 30,100 | 20,250 | 28,350 | 1.40 | 5.2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The diblock copolymers P3 and P4 were then used as components for the self-assembly of CCS polymers. These were prepared by mixing equimolar solutions of P3 and P4 in THF in the presence of TFA catalyst at a range of different concentrations (0.5–5.0 wt %), and the resulting solutions were left to equilibrate overnight before analysis (Table 2) by gel permeation chromatography–multiangle laser light scattering (GPC–MALLS) which allows the measurement of Mw and the radius of gyration (Rg). Online dynamic light scattering measurements also furnished the hydrodynamic radius (Rh). No macroscopic gelation was observed in any of these experiments, an observation which suggests the ‘inert’ blocks are successfully preventing macroscopic cross-linking of the polymer chains at the concentrations studied. All GPC traces (Fig. 3) display26 a disappearance of the peaks at ∼14 min corresponding to diblock copolymers P3 and P4, and the appearance of a major peak at lower elution volumes indicative of the formation of high molecular weight aggregates. These experiments indicate a dependence (Fig. 4) of the Mw of the CCS polymers formed upon the concentrations of the starting diblock polymers used, indicating CCS polymers of a desired size can be prepared simply by altering the concentration of polymer building blocks in the self-assembly reaction.
| Experiment number | Total diblock copolymer wt (%) | M n (g mol−1) | M w (g mol−1) | PDIa(Mw/Mn) | Average number of polymer chains per assemblyc | R g (nm) | R h (nm) | Structure sensitive ρ parameter (Rg/Rh) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a As determined by online static light scattering in THF (1.0 mL min−1) using experimentally determined dn/dc value (0.204 mL g−1). b As determined by online dynamic light scattering in THF (1.0 ML min−1). c Calculated by dividing Mw for CCS polymers by the average Mw of P3 and P4. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | 0.5 | 379,600 | 399,600 | 1.05 | 11 | 13.3 | 11.7 | 1.14 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 1 | 493,000 | 515,100 | 1.04 | 14 | 13.1 | 12.5 | 1.05 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 2 | 967,000 | 1,175,000 | 1.22 | 33 | 19.5 | 14.8 | 1.32 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | 3 | 1,337,000 | 1,651,000 | 1.23 | 46 | 27.7 | 17.7 | 1.56 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | 4 | 2,401,000 | 3,034,000 | 1.26 | 85 | 44.9 | 26.0 | 1.73 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | 5 | 5,235,000 | 6,808,000 | 1.30 | 190 | 61.9 | 34.4 | 1.80 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
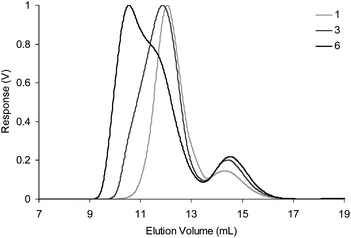 | ||
| Fig. 3 Differential refractive index (dRI) GPC traces for experiments 1, 3 and 6 in THF (1.0 mL min−1). Traces for experiments 2, 4 and 5 have been omitted for clarity. | ||
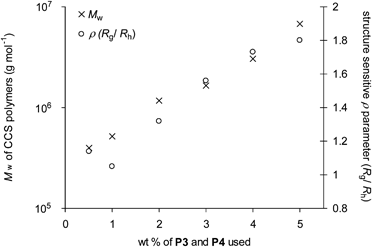 | ||
| Fig. 4 The dependence of Mw and ρ of the CCS polymers obtained from cross-linking equimolar amounts of polymers P3 and P4 at different concentrations. | ||
The Mw measurements as determined by GPC-MALLS were consistently larger than those determined by calibration against polystyrene standards, an observation consistent with the character of CCS polymers, which are more compact than their linear counterparts with the same molecular weights, suggesting that the CCS polymers prepared possess a cross-linked branched structure. Further evidence supporting the CCS polymer architecture can be obtained from the structure sensitive ρ parameter (ρ = Rg/Rh) defined by Burchard and co-workers.27 Those CCS polymers prepared at lower concentrations (0.5 and 1.0 wt %, Table 2, entries 1–2) possess ρ values ∼1.1, which is consistent28 with monodisperse regular star architectures and suggests that these lower concentrations are the optimum to form the most monodisperse species. The upward trend (Fig. 4) in ρ values for those CCS polymers prepared at higher concentrations (Table 2, entry 3–6) indicates increasing polydispersity, which maybe as a consequence of a degree of star-star couplings.
To investigate whether the CCS polymers are prone to requilibriation, solutions of CCS polymers containing sufficient TFA to ensure trans-imination could occur were stored for one week at concentrations lower and higher than the concentration at which they were prepared. Subsequent GPC analysis showed no change suggesting that the CCS polymers formed are kinetically very stable and do not ‘shrink’ or ‘grow’ in response to concentration changes.
Interestingly, all dRI GPC traces displayed the presence of a minor species eluting after ∼14.5 min. An analytical sample of this species was isolated by GPC and analyzed by matrix-assisted laser desorption ionization–time of flight (MALDI–TOF) mass spectrometry (Fig. 5). The mass spectrum suggests the presence of an aggregate with a molecular weight of approximately 60–65 kDa, suggesting strongly the formation of a 2–armed CCS polymer formed by the cross-linking between a single P3 chain and a single P4 chain. The mass spectroscopic evidence is, to the best of our knowledge, arguably the most convincing to date supporting the formation of 2-armed CCS polymers as a by-product during the formation of CCS polymers. Based on GPC chromatograms, Qiao and co-workers have postulated29 the formation of 2-armed CCS polymers as a minor component of polymers prepared through so-called ‘arm-first’ methods10 employing irreversible cross-linking of the polymer chains.
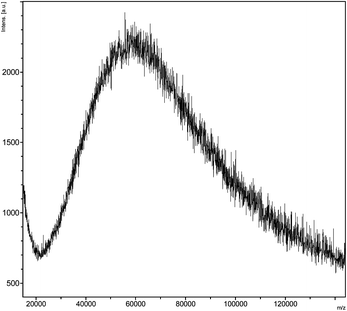 | ||
| Fig. 5 The MALDI-TOF mass spectrum of the minor product of polymer cross-linking, which suggests this species is a 2–arm core cross-linked star polymer. | ||
Further analysis of the GPC traces for cross-linking experiments 1–6 (Fig. 3 and ESI,† Fig. S4) also reveals a concentration dependence for the 2-armed species, as it appears that increased concentrations of 2-armed species are formed at higher initial concentrations of polymers P3 and P4. This concentration dependence suggests that the cross-linking of polymers P3 and P4 to form multi- and 2-armed species proceeds under kinetic control. It is likely that the formation of multi-armed species is a slower process than the formation of the 2-armed species simply because larger numbers of polymer chains are required to aggregate, and thus their concentration will decrease relative to the 2-armed species in the product distribution at higher starting concentrations.
To demonstrate the potential of these CCS polymers to undergo structural reconfiguration, a large excess of propylamine was added to a solution of CCS polymers (as prepared in Table 1, experiment 3) and the resulting mixture left to equilibrate overnight before GPC analysis (Fig. 6). The chromatogram obtained displayed the loss of the peak at ∼11.5 min corresponding to CCS polymers and the appearance of a peak at ∼14.0 min which corresponds to diblock copolymers. This observation confirms that all the imine bonds present within the CCS polymer have undergone trans-imination reactions, resulting in disassembly of the CCS polymer species and the generation of P4 and a derivative of P3 where all aldehydes are have transformed into propylimines. Alkyl imines are thermodynamically more stable than aniline imines, providing a driving force for trans-imination to occur. We do not envisage that the products of the CCS polymer disassembly can easily be reconverted back into CCS polymers, as this transformation would effectively require the removal of propylamine from the system. The ability to induce a significant change in polymer assembly architecture is potentially very useful because dramatic structural reconfiguration is likely to be accompanied by a significant change in the properties of the polymer, e.g. mechanical strength or viscosity.
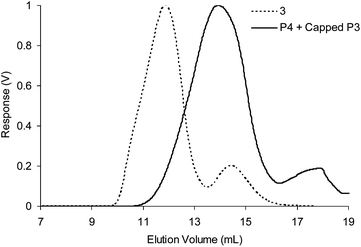 | ||
| Fig. 6 Differential refractive index (dRI) GPC traces indicating the decomposition of the CCS polymers prepared in Table 1 experiment 3 (dashed line) into block copolymers (solid line) after the addition of propylamine. | ||
The importance of the “inert” block in the formation of discrete CCS polymers was emphasized by a cross-linking experiment involving polymers P1 and P2b which do not possess “inert” blocks. Equimolar solutions of P1 and P2b in THF were combined in the presence of TFA catalyst at either 0.5 or 5 wt % and left to equilibrate. After approximately 15 min (5 wt %) or 2 h (0.5 wt %) a gel-like material was obtained (ESI,† Fig. S12) suggesting the formation of a macroscopic cross-linked gel. This gel could potentially hold promise as a so-called covalently adaptable network30 (CAN) constituting a new class of materials possessing adaptive and responsive properties on account of their dynamic imine cross-links.
Results from our experiments suggest that an ‘inert’ block is required to promote the formation of CCS polymer species, and that in the absence of an inert block the polymer chains will cross-link to form macroscopic gels. The propensity of polymers P1 and P2b to cross-link into gels, however, may also be as a consequence of the relatively high density of aldehyde and amino groups within the polymer. Work by the group of Otsuka and Takahara suggests18 that polymer chains without ‘inert’ blocks can cross-link to form spherical polymer-based nanogel architectures. This work utilizes cross-linking through a radical cross-over reaction, and the reactive functional group density within these polymers is much lower than the 1:1 ratio of amine/aldehyde:styrene used in our own polymers P1 and P2b. We therefore set out to investigate if it would be possible to form nanogel architectures by cross-linking polymer chains through imine bonds polymer chains when the reactive functional group density is lowered to 1:8.
Methyl methacrylate-based cross-linked nanogels
Attempts to utilize RAFT polymerizations to prepare analogues of copolymers P1 and P2b possessing different functional group densities were unsuccessful, affording copolymers whose compositions differed from the feed ratio when this value deviated from 1:1 of styrene: 1/2. Although the reasons for this observation are uncertain, the differences in reactivity of the monomers are a possible source of the problem. We therefore chose to focus on methacrylate-based copolymers prepared by copolymerization of the monomers M1 or M2 (Scheme 2) with methyl methacrylate. The aldehyde or amine functional groups within these monomers are located sufficiently far away from the vinyl groups to ensure these monomers possess similar reactivity to methyl methacrylate, thus ensuring that co-polymerizations can be controlled and the ratios of the two monomers within the resulting copolymers accurately reflected the feed ratios. The monomer M1 was prepared (Scheme 2) by O-alkylation of 4-formylphenol to afford the alcohol 3 which was then acylated with methacryloyl chloride. Monomer M2 was prepared by Boc-protection of 4-aminophenol to afford 4, which was then O-alkylated to furnish the alcohol 5. Subsequent acylation with methacryloyl chloride afforded the monomer M2. All synthetic steps to prepare these monomers proceed with high yields and were experimentally straightforward, allowing significant quantities to be prepared quickly.
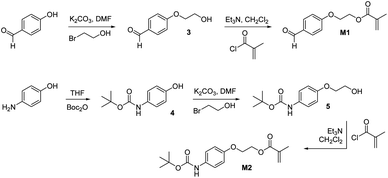 | ||
| Scheme 2 Synthesis of aldehyde and amino-functionalized methyl methacrylate-based monomers M1 and M2. | ||
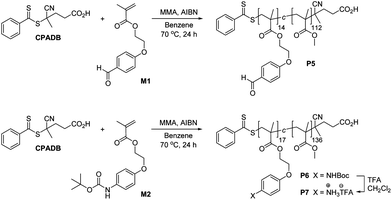
RAFT Polymerization was used to prepare (Scheme 3) the functionalized copolymers P5–P6. The RAFT chain transfer agent 4-(4-cyanopentanoic acid)dithiobenzoate31 (CPADB) was used to mediate the copolymerization of a 8: 1 mixture of methyl methacrylate: M1 in benzene at 70 °C to afford P5. Likewise, the copolymer P6 was prepared via RAFT polymerization of a 8: 1 mixture of methyl methacrylate: M2 in benzene at 70 °C, which was then treated with TFA/CH2Cl2 to remove the protecting groups to yield P7. Polymers P5–P7 were characterized (Table 3) by 1H NMR spectroscopy and GPC. All copolymers prepared possessed compositions identical to the feed ratios of the monomers used, and GPC analysis (Fig. 7) confirmed mono-modal distributions and low PDIs, suggesting the polymerizations proceed with a good degree of control. These observations also suggest that the functional monomers M1 and M2 are of similar reactivity to methyl methacrylate and the distribution of the monomers along the copolymers are likely to be random. The number average molecular weight (Mn) of polymers P5–P7 obtained by 1H NMR spectroscopy are not consistent with the Mn values determined by GPC although this discrepancy is probably on account of the relative structural differences between the functionalized copolymers P5–P7 and the poly(methyl methacrylate) standards used for GPC calibration. The values of Rh are consistent with linear polymer chains and indicate that there is no aggregation between the polymers when dissolved in THF.
 | ||
| Scheme 3 Synthesis of aldehyde and amine functionalized copolymers P5–P7. | ||
| Polymer | M n (g mol−1) | M n (g mol−1) | M w (g mol−1) | PDIb(Mw/Mn) | R h (nm) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a As determined by 1H NMR spectroscopy. b As determined by gel permeation chromatography in THF (1.0 mL min−1) calibrated against poly(methyl methacrylate) standards. c As determined by online dynamic light scattering measurements. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P5 | 14,750 | 19,200 | 23,500 | 1.23 | 3.5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P6 | 19,350 | 34,600 | 46,200 | 1.34 | 5.4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P7 | 19,600 | 30,750 | 41,600 | 1.35 | 5.9 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
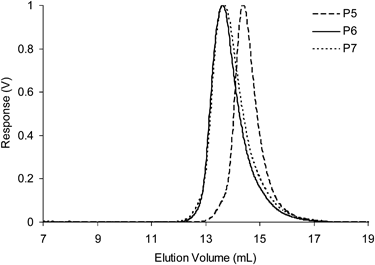 | ||
| Fig. 7 Differential refractive index (dRI) GPC traces (in THF at 1.0 mL min−1) of functional copolymers P5–P7. | ||
To investigate the formation of nanogels, equimolar solutions of P5 and P7 were mixed in THF and allowed to crosslink. A series of experiment (7–12, Table 4) were conducted over a range of concentrations (0.1–2 wt % of total copolymer building blocks). Solutions were left to equilibrate for 24 h before analysis by GPC-MALLS (Table 4) to obtain the measurement of Mn, Mw and Rg. Online dynamic light scattering measurements also furnished Rh. No macroscopic gelation was observed in any of these experiments, suggesting the formation of discrete nanogels. All GPC traces (Fig. 8) show a disappearance of peaks at ∼13.5–14.5 min corresponding to diblock copolymers P5 and P7 and the appearance of a major peak at lower elution volume indicating the formation of high molecular weight species. As with the styrenic CCS polymers, these experiments indicate (Fig. 9) a concentration dependence, with the Mw of the nanogels formed dependent upon the initial concentration of the copolymer building blocks used during cross-linking. The Mw measurements as determined by GPC-MALLS were consistently larger than those determined by calibration against linear polymethyl methacrylate standards, which suggests the nanogels formed are compact and their branched nature reduces their volumes, subsequently increasing their GPC elution volumes. Further evidence for the formation of nanogel architectures can again be obtained from the structure sensitive ρ parameter (ρ = Rg/Rh). Those nanogels formed at lower concentrations (experiments number 7–8) possess ρ values <0.8 which are consistent28 with compact spherical shapes. The upward trend (Fig. 9) in ρ values for those nanogels formed at higher concentrations (experiments 9–12) may indicate the presence of a degree of sphere-sphere couplings. These results suggest that cross-linking is best performed at low concentrations to ensure the formation of nanogels possessing low polydispersity. More importantly, these results also indicate that ‘inert’ blocks are not required for the formation of discrete nanogels by imine formation when the density of the functional groups involved in cross-linking is sufficiently low.
| Experiment number | Total copolymer wt (%) | M n (g mol−1) | M w (g mol−1) | PDIa (Mw/Mn) | Average number of polymer chains per assemblyc | R g (nm) | R h (nm) | Structure sensitive ρ parameter (Rg/Rh) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a As determined by online static light scattering in THF (1.0 mL min−1) using experimentally determined dn/dc value (0.095 mL g−1). b As determined by online dynamic light scattering in THF (1.0 mL min−1). c Calculated by dividing Mw for nanogels by the average Mw of P5 and P7. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | 0.1 | 777,600 | 797,400 | 1.03 | 24 | 8.8 | 12.8 | 0.69 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | 0.25 | 2,299,000 | 2,385,000 | 1.04 | 73 | 11.9 | 15.5 | 0.77 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | 0.5 | 4,870,000 | 5,515,000 | 1.13 | 169 | 18.7 | 21.0 | 0.89 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | 1 | 10,550,000 | 12,280,000 | 1.16 | 377 | 24.6 | 25.7 | 0.96 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 | 1.5 | 17,540,000 | 20,600,000 | 1.17 | 633 | 30.9 | 29.9 | 1.03 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12 | 2 | 23,335,000 | 27,620,000 | 1.18 | 849 | 39.0 | 34.4 | 1.13 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
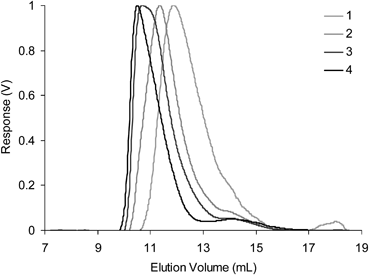 | ||
| Fig. 8 Differential refractive index (dRI) GPC traces (in THF at 1.0 mL min−1) for experiments 1–4. Traces for experiments 5 and 6 have been omitted for clarity as they posses similar elution volumes as experiment 4 suggesting that at high molecular weights the product assemblies are above the cut-off limit of the columns. | ||
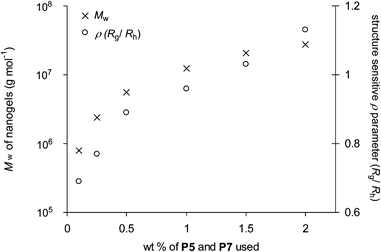 | ||
| Fig. 9 The dependence of Mw and ρ of the nanogels obtained by cross linking on the wt % of polymer P5 and P7 used. | ||
To investigate whether the nanogels are prone to requilibriation, solutions of nanogels containing sufficient TFA to ensure trans-imination could occur were stored for three days at concentrations lower and higher than the concentration at which they were prepared. Subsequent GPC analysis showed no change, suggesting that the nanogels are kinetically very stable and do not ‘shrink’ or ‘grow’ in response to concentration changes.
To gain insight into the kinetics of nanogel formation, experiment 5 (1.5 wt % of P5 and P7) was repeated by following the progress of the cross-linking by GPC-MALLS as a function of time (Fig. 10). Unfortunately the molecular weight cut-off of our GPC columns were too low to allow us to follow the entire self-assembly process by GPC, but online Rh measurements gave useful insight. The hydrodynamic radius was found to increase over time, reaching a plateau of around 30 nm after 6 h at which point equilibrium is presumably attained. The GPC measurements indicated that the component polymers P5 and P7 are consumed almost immediately. We postulate that those species which are formed initially from the cross-linking of P5 with P7 possess a small excess of aldehyde or amine functions, and that as the cross-linking continues these species can link together forming larger species until the system runs out of polymer building blocks and a constant hydrodynamic radius is observed. It is also possible that the ability of the imine bonds to undergo trans-imination processes also facilitates a certain amount of structural reorganization within the polymer assemblies.
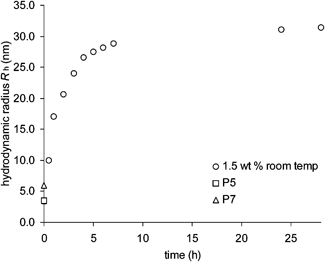 | ||
| Fig. 10 Nanogel formation over 24 h. The hydrodynamic radii of P5 and P7 are given for comparison. | ||
As it was possible for our styrenic CCS polymers to undergo component exchange in organic solvents, we envisaged that these nanogel species should also be able to undergo component exchange. To demonstrate this possibility, a large excess of ethanolamine was added to experiment 4 (1 wt % of P5 and P7) and the solution left to equilibrate overnight before GPC analysis. The chromatogram obtained (Fig. 11) displayed a loss of the peak at ∼11 min corresponding to nanogel species and the appearance of a peak at ∼14 min which corresponds to regenerated P7 and ethanolamine capped-P5. This observation confirms that all the imine bonds present within the nanogels have undergone trans-imination with ethanolamine resulting in the disassembly of the nanogels into single polymer chains.
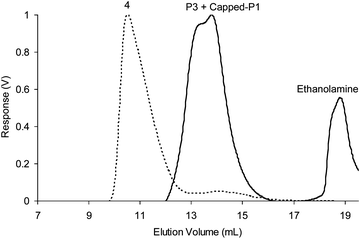 | ||
| Fig. 11 Differential refractive index (dRI) GPC traces (in THF at 1.0 mL min−1) displaying the disassembly of nanogels obtained from experiment 4 (1 wt % of P5 and P7). | ||
A series of cross-linking experiments were conducted at increased initial concentrations of P5 and P7 (3, 4 and 5 wt %). The solutions obtained at 3 and 4 wt % were very viscous and were not suitable for GPC analysis on account of the presence of small precipitates. Cross-linking at 5 wt % resulted in the formation of a macroscopic cross-linked organogel, confirmed by the vial inversion test (ESI,† Fig. S13). These results indicate that when cross-linking is performed at higher concentrations the formation of macroscopic networks is formed instead of discrete nanogels.
Conclusions
We have utilised RAFT polymerization to prepare novel aldehyde and amine functional styrenic- and methyl methacrylate-based copolymers and have demonstrated that these copolymers can cross-link through imine bond formation to prepare core cross-linked star polymers and spherical cross-linked nanogels. These polymer-based assemblies have been characterized by GPC-MALLS, which proved to be a very powerful tool in the analysis of these species. The potential for these assemblies to undergo structural reconfiguration has been shown through trans-imination with a small molecule amine, demonstrating the chemoresponsivness of these polymer systems. Functional copolymer building blocks have also be used to form macroscopic cross-linked organogel networks, where the presence of dynamic covalent imine linkages suggests that they may possess adaptive properties.A size dependence upon the initial concentration of copolymer building blocks was observed for both the formation of CCS and nanogel polymers, suggesting the self-assembly process is likely to be under kinetic control. We speculate that the reasons for this kinetic control are as a consequence of the relative stability of the imine bonds formed in organic solvents. Under the conditions of the self-assembly, imine bonds are reluctant to undergo hydrolysis back to their component amine and aldehyde reaction partners, presumably because of the low concentration of water in the reaction. Although it is likely that trans-imination reactions can occur after the polymer assemblies have formed, permitting them a very limited potential for reconfiguration, the fact that the imine cross-links cannot break means that significant structural reconfigurations also cannot occur. It would not be possible, for instance, for a single polymer chain to break all of its imine cross-links and leave the assembly. This contrasts with the CCS polymer system of Otsuka and Takahara, where the dynamic covalent linkages can be broken at elevated temperatures to a significant enough degree to permit a polymer chain to leave the cross-linked assembly, permitting far more scope for reconfigurations. Thus the lack of reversibility of the imine bond in organic solvents limits its usefulness in preparing adaptive and responsive species. The ability of the imine bond to readily undergo component exchange does, however, permit a level of chemoresponsive, as shown by the abilities of our cross-linked species to convert to linear polymer chains in the presence of an excess of amine. The imine bond becomes a far more interesting dynamic covalent reaction in water, as it is possible to alter significantly the position of its equilibrium with pH. We have recently shown32 how CCS polymers containing imine cross-links can undergo significant reconfigurations in water-soluble systems, and believe that in water the imine bonds hold far more potential for making structures which display responsive and adaptive abilities. We are now further investigating the use of imine bonds in the formation of CCS polymers, nanogels and macroscopic cross-linked gels in aqueous media.
Acknowledgements
Newcastle University (studentship to A.W.J) and One North East are gratefully thanked for support.Notes and References
- J.-M. Lehn, Prog. Polym. Sci., 2005, 30, 814–831 CrossRef CAS; J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14–27 CrossRef CAS.
- Y. Amamoto, Y. Higaki, Y. Matsuda, H. Otsuka and A. Takahara, J. Am. Chem. Soc., 2007, 129, 13298–13304 CrossRef CAS.
- (a) Z. Ge, J. Hu, F. Huang and S. Liu, Angew. Chem., Int. Ed., 2009, 48, 1798–1802 CrossRef CAS; (b) J.-H. Ryu, S. Jiwpanich, R. Chacko, S. Bickerton and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 8246–8247 CrossRef CAS.
- J. M. J. Paulusse and R. P. Sijbesma, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5445–5453 CrossRef CAS.
- T. F. Scott, A. D. Schneider, W. D. Cook and C. N. Bowman, Science, 2005, 308, 1615–1617 CrossRef CAS.
- D. S. Guo, S. Chen, H. Qian, H. Q. Zhang and Y. Liu, Chem. Commun., 2010, 46, 2620–2622 RSC.
- E. B. Murphy and F. Wudl, Prog. Polym. Sci., 2008, 33, 479–522 CrossRef.
- (a) K. Landfester and M. Antonietti, Colloids Colloid Assemblies, 2004, 175–215 CAS; (b) C. T. Black, C. B. Murray, R. L. Sandstrom and S. Sun, Science, 2000, 290, 1131–1134 CrossRef CAS; (c) J. W. Taylor and M. A. Winnik, J. Coat. Technol. Res., 2004, 1, 163–190 CrossRef CAS; (d) J. K. Oh, R. Drumright, D. J. Siegwart and K. Matyjaszewski, Prog. Polym. Sci., 2008, 33, 448–477 CrossRef CAS; (e) Z. L. Tyrrell, Y. Q. Shen and M. Radosz, Prog. Polym. Sci., 2010, 35, 1128–1143 CrossRef CAS.
- (a) H. Gao and K. Matyjaszewski, Prog. Polym. Sci., 2009, 34, 317–350 CrossRef CAS; (b) A. Blencowe, J. F. Tan, T. K. Goh and G. G. Qiao, Polymer, 2009, 50, 5–32 CrossRef CAS.
- A. V. Kabanov and S. V. Vinogradov, Angew. Chem., Int. Ed., 2009, 48, 5418–5429 CrossRef CAS.
- J. T. Wiltshire and G. G. Qiao, Aust. J. Chem., 2007, 60, 699–705 CrossRef CAS.
- K. Fukukawa, R. Rossin, A. Hagooly, E. Pressly, J. Hunt, B. Messmore, K. Wooley, M. Welch and C. J. Hawker, Biomacromolecules, 2008, 9, 1329–1339 CrossRef CAS.
- (a) T. Terashima, M. Kamigaito, K.-Y. Baek, T. Ando and M. Sawamoto, J. Am. Chem. Soc., 2003, 125, 5288–5289 CrossRef CAS; (b) B. Helms, S. J. Guillaudeu, Y. Xie, M. McMurdo, C. J. Hawker and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2005, 44, 6384–6387 CrossRef CAS; (c) T. Terashima, M. M. Ouchi, T. Ando, M. Kamigaito and M. Sawamoto, Macromolecules, 2007, 40, 3581–3588 CrossRef CAS.
- J. K. Oh, R. Drumright, D. J. Siegwart and K. Matyjaszewski, Prog. Polym. Sci., 2008, 33, 448–477 CrossRef CAS.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef.
- References on Living Radical Polymerisations: RAFT (a) G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS; (b) S. Perrier and P. Takolpuckdee, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5347–5393 CrossRef CAS; (c) G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 59, 669–692 CrossRef CAS: ATRP (d) K. Matyjaszewski and J. Xai, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS: NMP (e) C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS.
- Y. Higaki, H. Otsuka and A. Takahara, Macromolecules, 2006, 39, 2121–2125 CrossRef CAS.
- (a) Y. Amamoto, Y. Higaki, Y. Matsuda, H. Otsuka and A. Takahara, J. Am. Chem. Soc., 2007, 129, 13298–13304 CrossRef CAS; (b) Y. Amamoto, M. Kikuchi, H. Masunaga, S. Sasaki, H. Otsuka and Takahara, Macromolecules, 2010, 43, 1785–1791 CrossRef CAS.
- J.-H. Ryu, R. T. Chacko, S. Jiwpanich, S. Bickerton, R. P. Babu and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 17227–17235 CrossRef CAS.
- P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS.
- R. Manzotti, T. S. Reger and K. D. Janda, Tetrahedron Lett., 2000, 41, 8417–8420 CrossRef CAS.
- J. Jiang and S. Thayumanavan, Macromolecules, 2005, 38, 5886–5891 CrossRef CAS.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754–6756 CrossRef CAS.
- The shoulder present in the GPC trace for P4 is probably on account of the noticeable increase in viscosity of the polymerization reaction mixture, which may affect the kinetics of the polymerization process.
- dRI Traces and light scattering traces can display differences as the dRI response only depends on the concentrations of the molecules whereas the light scattering response depend on the molecular weights as well as concentrations. Complete sets of dRI and light scattering traces are presented in the ESI (S3, S4, S10 and S11).
- W. Burchard, M. Schmidt and W. H. Stockmayer, Macromolecules, 1980, 13, 1265–1272 CrossRef CAS.
- P. Lang, W. Burchard, M. S. Wolfe, H. J. Spinelli and L. Page, Macromolecules, 1991, 24, 1306–1314 CrossRef CAS.
- W. M. Spiniello, A. Blencowe and G. C. Qiao, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2422–2431 CrossRef.
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Macromolecules, 2010, 43, 2643–2653 CrossRef CAS.
- Y. Mitsukami, M. S. Donovan, A. B. Lowe and C. L. McCormick, Macromolecules, 2001, 34, 2248–2256 CrossRef CAS.
- A. W. Jackson and D. A. Fulton, Chem. Commun., 2011, 47, 6807–6809 RSC.
Footnote |
| † Electronic Supplementary Information (ESI) available: 1H NMR spectra of M1, M2 and P3–P7. GPC MALLS and dRI traces of experiments 1–12. Photographs displaying gelation of P1 and P2b and gelation of P5 and P7. See DOI: 10.1039/c1py00261a/ |
| This journal is © The Royal Society of Chemistry 2011 |