Dixanthate-terminated poly(butylene terephthalate). A novel RAFT/MADIX agent for the synthesis of well-defined triblock copolymers resulting from consecutive step- and chain-growth polymerization processes†
Aurélie
Sandeau
ab,
Stéphane
Mazières
a,
Caroll
Vergelati
b,
Cécile
Corriol
b and
Mathias
Destarac
*a
aUniversité Paul Sabatier, Laboratoire Hétérochimie Fondamentale et Appliquée, UMR-CNRS 5069, Bât 2R1, 118, route de Narbonne, 31062, Toulouse Cedex 9, France. E-mail: destarac@chimie.ups-tlse.fr
bRhodia Opérations, Centre de Recherches et Technologies de Lyon, 85, rue des Frères Perret, 69192, Saint-Fons Cedex, France
First published on 26th August 2011
Abstract
Well-defined triblock copolymers comprising a poly(butylene terephthalate) (PBT) mid-block and three different hydrophobic blocks (poly(tert-butyl acrylate) P(t-BA), poly(n-butyl acrylate) P(n-BA) and poly(tert-butyl acrylamide) P(t-BAm)) were successfully prepared by the combination of step-growth and RAFT/MADIX polymerizations. Two different synthetic strategies were investigated for the preparation of O-ethylxanthate-terminated PBT RAFT/MADIX agent. Firstly α,ω-dihydroxy-PBT synthesized by a step-growth polymerization was transformed into the corresponding dixanthate-functionalized PBT according to a two-step procedure. An alternative approach was performed by using a hydroxyl-functional xanthate as chain stopper in a single step-growth polymerization process. In both cases, the presence of xanthate terminal groups was confirmed by NMR spectroscopy and MALDI-TOF mass spectrometry. The resulting PBT with xanthate end-groups was used as a macro-chain transfer agent for the RAFT/MADIX polymerization of n-BA, t-BA and t-BAm. The synthesis of well-defined PBT-based triblock copolymers was confirmed by SEC analysis.
Introduction
Macromolecular engineering by means of mechanistic transformations1 allows the synthesis of block copolymers that cannot be made by a single polymerization mode. For instance, many synthetic strategies for the production of block copolymers from different living and controlled/living polymerization methods were successfully developed and largely exemplified.2 In particular, reversible-deactivation radical polymerization (RDRP),3 previously known as controlled radical polymerization, could be combined with other types of controlled chain-growth processes including living anionic4–6 and cationic polymerizations,7 ring-opening metathesis polymerization,8 and chain-growth condensation polymerization,9–13 thereby yielding well-defined polymer architectures from consecutive living/controlled polymerizations.The synthesis of block copolymers comprising step-growth14 and vinyl polymers is rather difficult due to the very different nature of the respective polymerization mechanisms. Such combination requires the control of terminal, lateral or in-chain functionality of the polycondensate which can activate another type of polymerization. To this end, a broad range of possibilities can be envisioned with RDRP in the presence of polycondensates as macro-initiators. Most classes of step-growth polymers such as polysulfones,15–17 polyurethanes,18,19 polyamides,20,21 poly(ether ether ketone),22 polycarbonates23,24 and polyesters25–29 were already studied in this context.
Among polyesters, poly(10-hydroxydecanoic acid)29 (PHDA) and glycolized poly(ethylene terephthalate)26 (PETG) were used as atom transfer radical polymerization (ATRP) macroinitiators for the polymerization of styrene (S) or glycidyl methacrylate (GMA) leading to PHDA-b-PS, PS-b-PHDA-b-PS, PS-b-PETG-b-PS and (PS-stat-PGMA)-b-PETG-b-(PS-stat-PGMA) copolymers. Poly(N-isopropyl acrylamide) was also grown from surface of PET films by surface-initiated ATRP.30 For all classes of step-growth polymerization, the employed techniques for the incorporation of the radical segment, such as polystyrene (PS), poly(methyl methacrylate), poly(n-butyl acrylate) (P(n-BA)) or poly(oligoethylene glycol methyl ether methacrylate) were mainly ATRP and nitroxide mediated polymerization (NMP). Surprisingly, reversible addition fragmentation chain transfer31/macromolecular design by interchange of xanthates32 (RAFT/MADIX) polymerization from polycondensate precursors bearing thiocarbonylthio end-groups was never reported. This undoubtedly represents an important synthetic challenge with great potential, since in contrast to the main other RDRP techniques, the RAFT/MADIX process is known for its excellent functional group tolerance and offers the advantage of controlling the polymerization of all the main classes of free-radically polymerizable monomers.33 More particularly, dithiocarbonate (also known as xanthate) RAFT/MADIX agents are suited to control not only the polymerization of conjugated monomers like acrylate and acrylamido monomers,34 but also that of nonconjugated monomers like vinyl esters,35 vinyl lactams36 and diallyl monomers.37 Advantageously, the chemical transformation of telechelic polymers into xanthate derivatives was already reported and has allowed the incorporation of well-defined hydrophilic polymers (e.g. poly(N-vinyl pyrrolidone) or poly(N-isopropyl acrylamide)) into polysiloxane,38,39 poly(ethylene glycol)40 or poly(ε-caprolactone)41 chains. Monoxanthate-terminated poly(ethylene-co-butylene) P(E-co-But) prepared by a two-step modification procedure of the corresponding hydroxy-terminated P(E-co-But) was also used as precursor for the RAFT/MADIX polymerization of vinyl acetate.42 Among the numerous classes of industrial step-growth polymers, PBT is one of the most important thermoplastic polyester covering a wide range of properties and applications.43 In order to improve the miscibility and optimize the end-use properties of polymer blends containing PBT,44 well-defined block copolymers comprising a PBT segment could act as efficient compatibilizers.
We report herein the controlled synthesis and characterization of well-defined triblock copolymers of poly(butylene terephthalate) (PBT) resulting from the combination of step-growth and RAFT/MADIX polymerizations. PBT with xanthate end-groups was first prepared by two synthetic strategies (Scheme 1). A first method has required the preparation of α,ω-dihydroxy PBT and its transformation according to a two-step nucleophilic substitution procedure. In the second approach a hydroxyl-functional xanthate chain stopper was directly added to the step-growth polymerization process. The resulting α,ω-dixanthate- functional PBT was then used as a macro-chain transfer agent for the RAFT/MADIX polymerization of n-butyl acrylate (n-BA), tert-butyl acrylate (t-BA) and tert-butyl acrylamide (t-BAm).
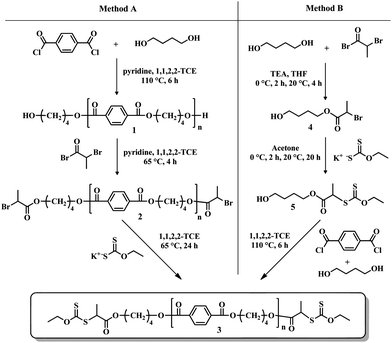 | ||
| Scheme 1 Synthetic strategies for O-ethyl xanthate-telechelic poly(butylene terephthalate). | ||
Experimental
Materials
The 2,2′-azobis(isobutyronitrile) initiator (AIBN, 98%) was purchased from Janssen Chimica and recrystallized three times from methanol before use. Other reagents were used as received without further purification. 1,1,2,2-tetrachloroethane (1,1,2,2-TCE, >98%) was purchased from Fluka. 1,4-butanediol (BTDiOH, 99%), terephthaloyl chloride (TPhCl, 99%), 2-bromopropionyl bromide (97%) and O-ethyl xanthic acid potassium salt (96%) were purchased from Sigma-Aldrich. n-butyl acrylate (n-BA, 99%), tert-butyl acrylate (t-BA, 99%) and tert-butyl acrylamide (t-BAm, 99%) were supplied from Acros Organics.Characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 vol.%) as the eluent at 35 °C with a flow rate of 1 mL min−1. The system was calibrated by using narrow polystyrene standards ranging from 970 to 117000 g mol−1.
:2:0.25. 2 μL of sample mixture was spotted on the MALDI-TOF MS sampler holder and slowly dried to allow matrix crystallization. A deisotoping program (Polymerix software) was used to facilitate the peak-assignment process by removing the contributions of the M + 1 and M + 2 isotopic peaks.
:7 vol.%) as the eluent at 35 °C with a flow rate of 1 mL min−1. The system was calibrated by using narrow polystyrene standards ranging from 970 to 117000 g mol−1.
:2:0.25. 2 μL of sample mixture was spotted on the MALDI-TOF MS sampler holder and slowly dried to allow matrix crystallization. A deisotoping program (Polymerix software) was used to facilitate the peak-assignment process by removing the contributions of the M + 1 and M + 2 isotopic peaks.
Syntheses
Method A – Step-growth polymerization following by end-group transformation reactions.
Synthesis of dihydroxy-terminated poly(butylene terephthalate) (1). PBT 1 was prepared following a modified protocol from Boutevin et al.46 1,4-Butanediol (7.90 g, 87.8 mmol) and pyridine (8.40 g, 106.3 mmol) were simultaneously added under argon to a solution of terephthaloyl chloride (15.03 g, 74.1 mmol) in 1,1,2,2-TCE (120 mL) at room temperature in a 250 mL three-neck round-bottom flask equipped with a magnetic stirrer and a reflux condenser. The reaction mixture was heated under argon at 110 °C for 6 h. The solution was then cooled to ambient temperature. PBT 1 was precipitated from the solution into a large excess of absolute acetone and isolated by filtration. The resulting solid was then washed with water and dried under vacuum for 24 h at 60 °C. (12.5 g, yield: 77%). Mn,NMR = 2100 Da, Mn,SEC = 3200 Da, Đ = 1.36.
1H NMR (300 MHz, C2D2Cl4): δ (ppm) 8.14 (s, C6H4), 4.46 (s, -CH2-OCO- of the repeating unit), 4.41–4.39 (t, -CH2-OCO- of the terminal unit), 3.77–3.73 (t, HO-CH2- of the terminal unit), 2.00 (s, -CH2-CH2-OCO- of the repeating unit), 1.95–1.88 (m, HO-CH2-CH2- of the terminal unit), 1.81–1.72 (m, -CH2-CH2-OCO- of the terminal unit). 13C NMR (75 MHz, C2D2Cl4): δ (ppm) 165.74 (-OCO-), 134.42 (Cipso (C6H4)), 130.09 (Co,m (C6H4)), 65.38 (-CH2-OCO- of the terminal unit), 64.98 (-CH2-OCO- of the repeating unit), 62.39 (HO-CH2- of the terminal unit), 29.34 (HO-CH2-CH2- of the terminal unit), 25.64 (-CH2-CH2-OCO- of the repeating unit), 25.42 (-CH2-CH2-OCO- of the terminal unit).
Synthesis of dibromopropionate-terminated PBT (2). PBT 1 (12 g, 5.8 mmol) was dissolved in 120 mL of 1,1,2,2-TCE at 110 °C in a 250 mL two neck round-bottom flask equipped with an addition funnel. The temperature was decreased to 65 °C. At this temperature, pyridine (1.2 g, 15.2 mmol) was first introduced and a solution of 2-bromopropionyl bromide (3.8 g, 17.7 mmol) in 5 mL of 1,1,2,2-TCE was added dropwise under argon for 30 min. The homogeneous reaction mixture was stirred under argon at 65 °C for 4 h, and then cooled to ambient temperature. PBT 2 was precipitated from the solution into a large excess of absolute acetone, filtered and washed with water. The obtained solid was then dried under vacuum for 24 h at 60 °C. (12 g, yield: 86%). Mn,NMR = 2350 Da, Mn,SEC = 3300 Da, Đ = 1.37.
1H NMR (300 MHz, C2D2Cl4): δ (ppm) 8.14 (s,C6H4), 4.46 (s, -CH2-OCO- of the repeating unit), 4.42–4.39 (q, -CH-CH3- of the terminal unit), 4.32–4.23 (q, -CH2-OCO-CH- of the terminal unit), 2.00 (s, -CH2-CH2-OCO- of the repeating unit), 1.90 (s, -CH2-CH2-OCO- of the terminal unit), 1.87–1.85 (d, -CH-CH3- of the terminal unit). 13C NMR (75 MHz, C2D2Cl4): δ (ppm) 165.87 (-OCO-), 134.06 (Cipso (C6H4)), 129.70 (Co,m (C6H4)), 65.56 (-CH2-OCO-CH- of the terminal unit), 65.10 (-CH2-OCO- of the repeating unit), 40.64 (-CH-CH3- of the terminal unit), 25.55 (-CH2-CH2-OCO- of the repeating unit), 21.80 (-CH-CH3- of the terminal unit).
Synthesis of xanthate-end-functionalized poly(butylene terephthalate) (3). PBT 2 (12 g, 5.11 mmol) was dissolved in 120 mL of 1,1,2,2-TCE at 65 °C in a round-bottom flask and O-ethyl xanthic acid potassium salt (2.5 g, 15.6 mmol) was added portionwise for 30 min. The reaction mixture was stirred at 65 °C for 24 h and cooled to ambient temperature. PBT 3 was precipitated in absolute acetone, filtered and washed with water. The resulting solid polymer was dried under vacuum for 24 h at 60 °C. (11.6 g, yield: 93%), Mn,NMR = 2450 Da, Mn,SEC = 3500 Da, Đ = 1.32.
1H NMR (300 MHz, C2D2Cl4) δ (ppm) 8.14 (s, C6H4), 4.69–4.62 (q, CH3-CH2-O- of the terminal unit), 4.44 (s, -CH2-OCO- of the repeating unit), 4.24 (t, -CH2-OCO-CH- of the terminal unit), 2.00 (s, -CH2-CH2-OCO- of the repeating unit), 1.89 (s, -CH2-CH2-OCO- of the terminal unit), 1.62–1.60 (d, -CH-CH3 of the terminal unit), 1.46–1.41 (t, -O-CH2-CH3 of the terminal unit). 13C NMR (75 MHz, C2D2Cl4) δ (ppm) 165.87 (-OCO-), 134.07 (Cipso (C6H4)), 129.71 (Co,m (C6H4)), 70.69 (-O-CH2-CH3 of the terminal unit), 65.10 (-CH2-OCO- of the repeating unit), 47.26 (-CH-CH3 of the terminal unit), 25.43 (-CH2-CH2-OCO-), 17.01 (-CH-CH3 of the terminal unit), 13.89 (-O-CH2-CH3 of the terminal unit).
Method B – Step-growth polymerization in the presence of a hydroxyl-functional xanthate as chain-stopper.
Synthesis of hydroxybutyl-2-bromopropionate (4). 4 was prepared by taking inspiration from the syntheses of bromo iniferter analogs.6,47 1,4-Butanediol (200 g, 2,2 mol) and triethylamine (TEA, 40 g, 0.40 mol) were dissolved in dry tetrahydrofurane (THF, 400 mL) in a 1 L round-bottom flask equipped with a magnetic stirrer and an addition funnel. 2-Bromopropionyl bromide (23.9 g, 0.11 mol) was dissolved in 20 mL of dry THF and the solution was added dropwise under argon to the reaction mixture over 2 h at 0 °C. After an additional stirring of 20 h at room temperature, the triethylammonium salts were filtered, THF was evaporated and the residual product was dissolved in dichloromethane and washed three times with water. The crude product was purified by preparative high performance liquid chromatography on an apparatus equipped with a Prochrom column (internal diameter = 50 mm, 250 g of silica 10 μm) and a preparative UV detector at a wavelength of 254 nm. A mixture of petroleum ether : ethyl acetate (65
:35 vol.%) was used as the eluent with a flow-rate of 70 mL min−1. 4 was obtained as a yellow oil and analyzed by GC-MS. (18.4 g, yield: 74%).
1H NMR (300 MHz, CDCl3) δ (ppm) 4.39–4.32 (q, 1H, -CH-CH3), 4.23–4.12 (dd, 2H, -CH2-OCO-), 3.67–3.63 (t, 2H, HO-CH2-), 2.41 (s, 1H, -OH), 1.81–1.80 (d, 3H, -CH-CH3), 1.78–1.70 (m, 2H, -CH2-CH2-OCO-), 1.69–1.59 (m, 2H, HO-CH2-CH2-). 13C NMR (75 MHz, CDCl3) δ (ppm) 170.39 (-O-CO-), 65.82 (-CH2-OCO-), 62.00 (HO-CH2-), 40.24 (-CH), 28.85 (HO-CH2-CH2-), 24.89 (-CH2-CH2-OCO-), 21.62 (-CH3). MS: m/z [M+˙] theo. 224.00, exp. 224.09.
Synthesis of S-(hydroxybutyl-2-propionyl) O-ethyl dithiocarbonate (5). Compound 4 (10 g, 44.4 mmol) was dissolved in 150 mL of acetone at room temperature. O-Ethyl xanthic acid potassium salt (9.5 g, 59.4 mmol) was added to the solution portionwise over 30 min at 0 °C. The solution was stirred at room temperature for 20 h. After filtration of the potassium bromide salts and evaporation of acetone, the product was extracted with diethyl ether. The excess of O-ethyl xanthic acid potassium salt was also eliminated by filtration. Compound 5 was obtained as a yellow oil after evaporation of the solvent and dried under vacuum. (12.5 g, yield: 95%). Product 5 was analyzed by GC-MS.
1H NMR (300 MHz, CDCl3) δ (ppm) 4.57–4.50 (q, 2H, -O-CH2-CH3), 4.31–4.24 (q, 1H, -CH-CH3), 4.10–4.05 (t, 2H, -CH2-OCO-), 3.55–3.51 (t, 2H, HO-CH2-), 2.94 (s, 1H, -OH), 1.70–1.62 (m, 2H, -CH2-CH2-OCO-), 1.60–1.52 (m, 2H, HO-CH2-CH2-), 1.51–1.50 (d, 3H, -CH-CH3), 1.36–1.30 (t, 3H, -O-CH2-CH3). 13C NMR (75 MHz, CDCl3) δ (ppm) 211.88 (-SCS-), 171.46 (-OCO), 70.30 (-O-CH2-CH3), 65.47 (-CH2-OCO-), 62.15 (HO-CH2-), 47.67 (-CH), 28.96 (1C, HO-CH2-CH2-), 25.04 (-CH2-CH2-OCO-), 16.59 (-CH3), 13.28 (-O-CH2-CH3). MS: m/z [M+˙] theo. 266.06, exp. 266.06.
Synthesis of dixanthate-end-functionalized poly(butylene terephthalate) (3). Terephthaloyl chloride (4.10 g, 20.2 mmol) was dissolved in 40 mL of 1,1,2,2-TCE at room temperature. 1,4-butanediol (1.48 g, 16.4 mmol) and compound 5 (2 g, 7.5 mmol) were simultaneously added under argon to the solution of terephthaloyl chloride. The reaction mixture was heated under argon at 110 °C for 6 h. After this period, the solution was cooled to ambient temperature. PBT 3 was precipitated in absolute acetone and filtered. The resulting solid was dried under vacuum for 24 h at 60 °C. (3.7 g, yield: 85%). Mn,NMR = 2350 Da, Mn,SEC = 3150 Da, Đ = 1.36.
Assignments of 1H and 13C NMR peaks are similar to compound 3 synthesized by method A. Spectra are available in the ESI† (Fig. S2 and S3).
This same procedure was applied for the polymerization of t-BA and t-BAm.
Results and discussion
The controlled incorporation of xanthate terminal groups in PBT is essential for the success of the following triblock copolymer synthesis by RAFT/MADIX polymerization. Two synthetic routes were investigated for the preparation of dixanthate end-functionalized PBT as shown in Scheme 1. Method A follows the most often used approach for the preparation of α,ω-telechelic macro-initiators through an end-group transformation chemistry.15,17,19 The hydroxyl end groups of PBT, obtained by a step-growth polymerization using an appropriate excess of diol monomer, were transformed into xanthates according a two-step nucleophilic substitution procedure. The second synthetic strategy (method B) employs a functional chain stopper 5 bearing the xanthate functionality and a hydroxyl group which is directly incorporated in the step-growth polymerization process to yield the xanthate-terminated PBT 3 in one single step.Both methods have allowed the access to α,ω-dixanthate- functional PBT capable of controlling the RAFT/MADIX polymerization of acrylates and acrylamido monomers.
Synthesis of a dixanthate-terminated poly(butylene terephthalate) RAFT/MADIX agent – Method A
A three-step synthetic route was used to prepare xanthate end-functionalized PBT as shown in method A of Scheme 1. Firstly, low molar mass α,ω-dihydroxy PBT 1 was obtained in a solution of 1,1,2,2-tetrachloroethane (1,1,2,2-TCE) at 110 °C by a step-growth polymerization of 1,4-butanediol (BTDiOH) and terephthaloyl chloride (TPhCl) in the presence of pyridine, with an appropriate excess of 1,4-butanediol ([BTDiOH]0/[TPhCl]0 = 1.19). The quantitative conversion of acyl chloride functions was confirmed by 1H and 13C NMR analysis with the extinction of the terephthaloyl chloride proton signals at 8.3 ppm and acyl chloride carbons at 167.9 ppm. The residual 1,4-butanediol was removed after precipitation of the polymer in acetone. The resulting dihydroxy-terminated PBT 1 was treated with a 3-fold excess of 2-bromopropionyl bromide in the presence of pyridine in 1,1,2,2-TCE at 65 °C for 4 h to yield the dibromopropionate-terminated PBT 2. The PBT-based RAFT/MADIX agent 3 was finally obtained via a reaction between the PBT 2 and an excess of O-ethyl xanthic acid potassium salt in 1,1,2,2-TCE at 65 °C for 24 h. Mn and dispersities (Đ = Mw/Mn) values for the three hydroxyl-, bromopropionate- and xanthate- functionalized PBTs were determined by SEC analysis using a chloroform : dichloroacetic acid mixture (93:7 vol.%) recommended by Shodex48 for PBT as the eluent at 35 °C with a PS calibration. Assuming that all the PBT chains were terminated by hydroxyl groups, the number-average molar mass Mn was also calculated by the comparison of the 1H NMR signals a at 8.12 ppm corresponding to the four aromatic protons of the repeating unit with d at 3.75 ppm related to the four methylene protons next to the oxygen of the BTDiOH terminal unit (Fig. 1 (1)). The SEC and 1H NMR analyses of the starting dihydroxy PBT (Mn,NMR = 2100 g mol−1, Mn,SEC = 3200 g mol−1, Đ = 1.36) and final dixanthate PBT (Mn,NMR = 2450 g mol−1, Mn,SEC = 3500 gmol−1, Đ = 1.32) showed similar Mn and unchanged Đ values, which revealed that no chain degradation occurred during the end-group modification reactions.
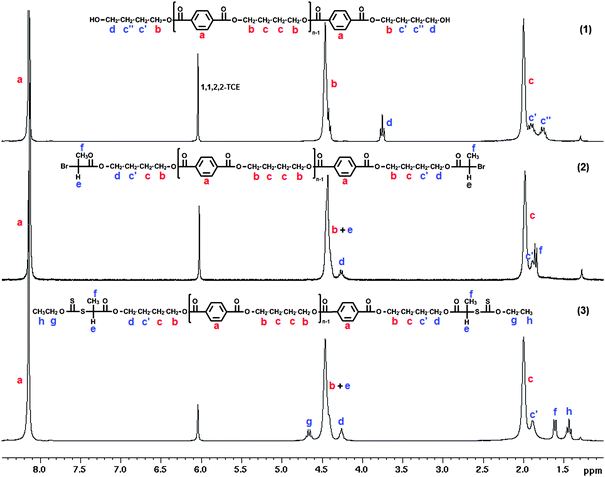 | ||
| Fig. 1 1H NMR spectra of (1) hydroxyl- (2) bromopropionate- and (3) xanthate-end-functionalized poly(butylene terephthalate). | ||
Quantitative transformation of hydroxyl to xanthate end groups was confirmed by 1H NMR spectroscopy (Fig. 1) and MALDI-TOF MS analyses (Fig. 2). In Fig. 1, signals a, b and c were attributed to the protons of butylene terephthalate units. After esterification (Fig. 1 (2)), the signal d at 3.75 ppm characteristic of the methylene groups in α-position of PBT hydroxyl end groups was shifted to higher field at 4.3 ppm and a doublet f assigned to the 3 protons of bromopropionate methyl group appeared at 1.8 ppm. The peak e of the methine proton on the α-carbon of the bromine was masked by the signal b. The last chain-end modification with the O-ethyl xanthate group was confirmed by the shifting of the doublet f from 1.8 ppm to 1.6 ppm and the appearance of signals g and h respectively at 4.6 and 1.4 ppm characteristic of the O-ethyl xanthate end groups.
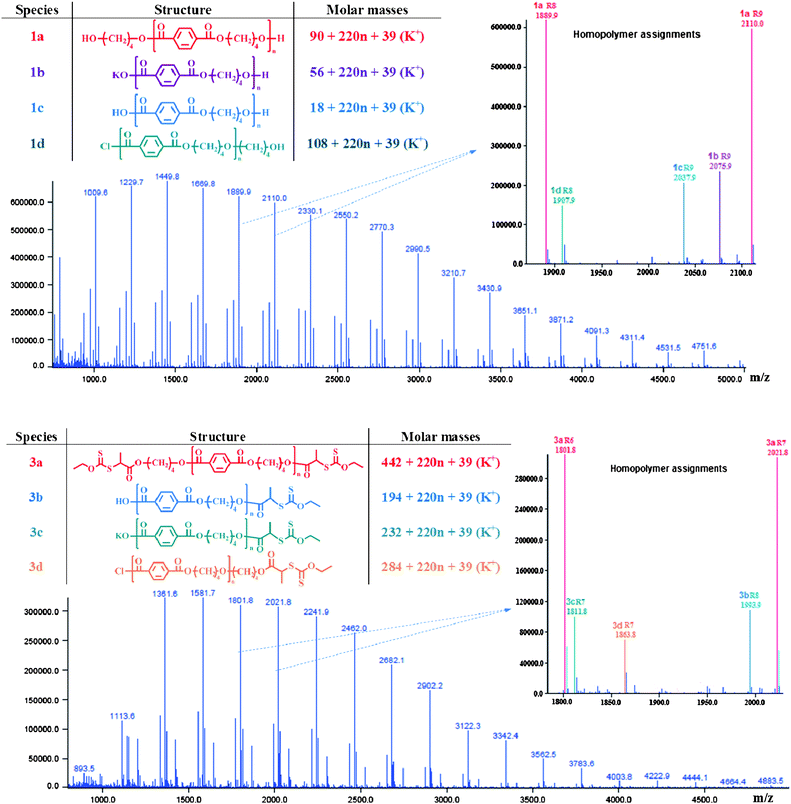 | ||
| Fig. 2 MALDI-TOF mass spectra of (1) hydroxyl- and (3) xanthate-end functionalized PBT after deisotoping procedure with structural homopolymers assignments of the peaks. | ||
A MALDI-TOF MS analysis was carried out to complete the characterization of the three telechelic PBTs. The spectra were obtained through the use of a dithranol matrix and trifluoroacetic acid potassium salt as the cationization reagent. HABA was also tested as the matrix due to its ability to analyse PBT samples49–51 but was not a suitable matrix for revealing bromopropionate and xanthate end groups in contrast to dithranol. Fig. 2 reports the MALDI-TOF spectra of the hydroxyl- and xanthate- functionalized PBTs (respectively 1 and 3) after the deisotoping procedure. Structural assignments of each mass distribution were performed by enlarging the spectra of 1 and 3 between two PBT repeating units. The two MALDI-TOF spectra contain four mass distributions in the range 1000–5000 Da with mass intervals corresponding to the molar mass of the PBT repeating unit (220 u). The most intense distribution of 1 at m/z = 90 + 220n + 39 corresponds to the expected PBT chains with hydroxyl groups at both ends (population 1a, Fig. 2). Second and third populations (respectively 1b and 1c in Fig. 2) appeared at m/z = 18 + 220n + 39 and m/z = 56 + 220n + 39 and could be assigned to the potassium adduct of PBT chains bearing the hydroxyl group at one end and COOK or terephthalic acid functionalities at the other end, respectively. The fourth mass distribution at m/z = 108 + 220n + 39 (population 1d, Fig. 2) was attributed to PBT chains with a hydroxyl end and the presence of an undesired ether linkage in the PBT segment as earlier reported.49 The comparison between the structural assignments of the peaks of the MALDI spectra of PBTs 1 and 3 confirmed that hydroxyl end groups of each species of 1 were transformed into xanthates, whereas the unreactive COOH and COCl groups which were identified in PBT 1 were still present in the final product 3. The MALDI-TOF MS analysis of 3 revealed the presence of the expected PBT chains with xanthate groups at both ends at m/z = 442 + 220n + 39 (species 3a, Fig. 2) together with three side populations (3b, 3c and 3d, Fig. 2) bearing the O-ethyl xanthate functionality at one end and one of the undesired COOH or COCl end-groups at the other end. The MALDI-TOF analysis confirmed the existence of PBT 3 with xanthate termini capable of activating the RAFT/MADIX polymerization.
Quantative compositional analysis cannot be obtained in MALDI-TOF MS due to the different ionization abilities of the polymer end-groups.52 Indeed, Puglisi et al.50 showed that the ionization of hydroxyl end groups was discriminated in comparison to the terephtalic acid end groups of PBT using HABA as the matrix. However, it is worth mentioning that the signals characteristic of terephthaloyl chloride protons at 8.3 ppm, acyl chloride carbons at 168 ppm and COOH carbon at 171 ppm were not detected by NMR spectrometry. Thus, although the MALDI-TOF MS analysis revealed that dixanthate-terminated PBT was contaminated by monoxanthate PBT, the concentration of the undesirable end-groups was low.
Synthesis of dixanthate-terminated poly(butylene terephthalate) RAFT/MADIX agent – Method B
Only a few research groups had recourse to a functional chain stopper in step-growth polymerization for the preparation of telechelic macro-initiators for RDRP.23,53 Block and graft copolymers of polycarbonate (PC) and polystyrene (PS) were prepared through the use of a chain stopper bearing the bromoisobutyryloxy functionality capable of activating the ATRP process at the end or side groups of the PC chain.23 Another approach for the synthesis of PS-b-PC-b-PS copolymers was the preparation of a hydroxy-functional PS by nitroxide mediated polymerization (NMP) which was added to step-growth polymerization involving bisphenol A and triphosgene.53In our case, the use of a hydroxy-xanthate chain stopper was a convenient way for the preparation of xanthate-terminated PBT in a one-step procedure. In contrast to method A, the transformation chemistry of hydroxyl into xanthate group was accomplished directly on the 1,4-butanediol monomer which was more environment-friendly in term of solvent and temperature. Indeed, the transformation reactions were operated at room temperature in THF and acetone while method A has required the use of 1,1,2,2-TCE at 65 °C due to the insolubility of PBT in usual solvents at room temperature.
The synthesis of the functional chain stopper was accomplished as follows: one of the two hydroxyl groups of 1,4-butanediol was transformed into xanthate functionality by two nucleophilic substitution reactions (Scheme 1). Firstly, 2-bromopropionyl bromide was added slowly to a large excess of 1,4-butanediol (20 equivalents) to form preferentially the expected compound 4 which was contaminated by the presence of the corresponding dibromo compound 4′. The relative molar proportion of 4 and 4′, determined by 1H NMR analysis, was governed by the molar ratio between 1,4-butanediol and 2-bromopropionyl bromide (see Fig. S1†). 10 equivalents of 1,4-butanediol were necessary to form more than 80% of hydroxybutyl-2-bromopropionate 4. There was no significant difference between 20 and 50 equivalents of 1,4-butanediol which led to the formation of 4′ in both cases. The purity of 4 is key for the preparation of the chain stopper. Indeed stoichiometry is the main parameter which governs DPn and the nature of end groups in step growth polymerization. The two compounds 4 and 4′ were isolated by preparative high performance liquid chromatography with a mixture of petroleum ether : ethyl acetate (65:35 vol.%) as the eluent. The purity of 4 was confirmed by GC-MS analysis. In a second step, the transformation of the bromopropionate group into xanthate functionality was readily accomplished in acetone with a small excess of O-ethyl xanthic acid potassium salt (1.3 equivalent). Quantitative conversion was confirmed by 1H NMR with the shifts of the –CH3 signals from 1.80 to 1.50 ppm and –CH from 4.35 to 4.28 ppm. The two signals characteristic of the O-ethyl xanthate group appeared at 4.53 (–OCH2CH3) and 1.33 ppm (–OCH2CH3). Purity of 5 was also confirmed by GC-MS analysis. In the last step, compound 5 was employed as chain stopper in the presence of BTDiOH and TPhCl for the direct synthesis of xanthate-terminated PBT in 1,1,2,2-TCE at 110 °C for 6 h. DPn and the nature of end groups were governed by the stoichiometric ratio between the difunctional monomers and the chain stopper according to the Carothers equation.54 Equality between the concentrations of acyl chloride and hydroxyl functions was required to obtain PBT chains with xanthate end groups at both ends. Depending on the molar ratio q between 5 and BTDiOH, several xanthate-terminated PBTs with different targeted DPn values were synthesized as shown in Table 1. Experimental DPn was determined by 1H NMR following the procedure explained in method A with the assumption that all the PBT chains were bearing the xanthate functionality at both ends. The Mn values of the resulting PBT homopolymers, similar to those obtained by SEC analysis (Table 1), increased with the reduction of the molar ratio q between the chain stopper and 1,4-butanediol in accordance to the Carothers equation. However, the difference between theoretical and experimental values and dispersities Đ increased when high values of DPn were targeted, which was characteristic of a gradual loss of control. NMR analysis of P1, P2, P3 and P4 were similar to those of the dixanthate-terminated PBT obtained from method A (see Fig. S2†, S3† for NMR spectra of P2).
| P | q | DP n(theory) | M n(theory) /g mol−1 | DP n(NMR) | M n(NMR) /g mol−1 | M n(SEC) /g mol−1 | Đ |
|---|---|---|---|---|---|---|---|
|
a
q = n5/2nBTDiOH.
b
DP
n (theory) = (2 + 3q)/q.
c
M
n (theory) = 220DPn (theory) + Mend-group.
d
M
n (NMR) = 220DPn (NMR) + Mend-group.
e
M
n and dispersities Đ determined by RI-SEC in a chloroform : dichloroacetic acid mixture (93:7 vol.%). Calibration by PS standards.
|
|||||||
| P1 | 0.38 | 8 | 2 261 | 7.7 | 2 136 | 2 300 | 1.27 |
| P2 | 0.24 | 12 | 2 972 | 8.7 | 2 356 | 3 150 | 1.36 |
| P3 | 0.18 | 14 | 3 567 | 10 | 2 642 | 3 400 | 1.43 |
| P4 | 0.12 | 19 | 4 666 | 12.5 | 3 192 | 3 550 | 1.55 |
| P5 | 0.05 | 43 | 9 831 | 26 | 6 162 | 5 050 | 1.64 |
A very small signal at 3.75 ppm characteristic of the methylene group next to the hydroxyl functionality was present in the 1H NMR spectrum while the signal of the corresponding carbon was not detected by 13C NMR. The MALDI-TOF spectra revealed the presence of four species with the most intense distribution corresponding to the PBT chains with xanthate end-groups at both ends (see Fig. S4†). Species 3b and 3c identified from the MALDI-TOF spectrum of PBTA ((3) in Fig. 2) were also present in the spectrum of PBTB. Only the fourth distribution differed from 3d of PBTA and was attributed to PBT chains bearing a xanthate functionality at one end and an hydroxyl end-group at the other end in accordance to 1H NMR analysis. These results confirmed the control of xanthate end-groups via method B.
Finally both methods A and B have allowed the access to well-controlled low molar mass α,ω-dixanthate PBTs. The presence of xanthate functionalities capable of activating the RAFT/MADIX process at each chain of the PBTs was confirmed by MALDI-TOF MS analysis. The xanthate-terminated PBTs coming from method A and one of those coming from method B (P2) were tested as macro-RAFT/MADIX transfer agents for the polymerization of some acrylate and acrylamido monomers.
Synthesis and characterization of PBT-based triblock copolymers by RAFT/MADIX polymerization
The two xanthate-capped PBT macro-transfer agents synthesized by methods A and B (PBTA and PBTB (P2) respectively) were used to prepare triblock copolymers with poly(n-butyl acrylate), P(n-BA) poly(tert-butyl acrylate) P(t-BA) and poly(tert-butyl acrylamide) P(t-BAm). RAFT/MADIX initiation and solvent conditions were chosen to ensure a good solubility of the PBT macro-xanthate transfer agent and nearly quantitative monomer conversions. Polymerizations were carried out in the presence of AIBN at 70 °C in 1,1,2,2-TCE. Preliminary studies have shown that transfer to 1,1,2,2-TCE had no noticeable influence on the control of Mn with targeted Mn of 10 000 g mol−1 for P(n-BA) and P(t-BA) homopolymers synthesized in the presence of a model xanthate for the PBT chain end of general structure CH3CH(CO2CH3)S(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S)OCH2CH3 (Rhodixan A1 supplied by Rhodia) (see Table S5 in the ESI†).
S)OCH2CH3 (Rhodixan A1 supplied by Rhodia) (see Table S5 in the ESI†).
In order to establish suitable SEC conditions for the analysis of both PBT precursor and acrylate-based triblock copolymers, a small proportion of dichloroacetic acid in chloroform (CHCl3 : CHCl2COOH = 93:7 vol.%) was found to be necessary to ensure a good solubility of the polymers. In contrast, all the PBTs of the study were found to be insoluble in pure chloroform. The solubility of the triblock copolymers was further studied by 1H NMR analysis (Table 2). Experimental molar fraction of PBT (xPBT exp) was determined by comparing two distinct 1H NMR signals characteristic of the protons of the repeating units of each block. It was shown that for all triblock copolymers, xPBT exp was lower in pure CDCl3 than in CDCl3:CHCl2COOH (see an example of 1H NMR spectra in Fig. S6†). The presence of P(t-BA), P(n-BA) and P(t-BAm) blocks in the copolymer allowed only partial solubilisation of the PBT block in pure deuterated chloroform, whereas xPBT in the CDCl3 : CHCl2COOH mixture was very close to the theoretical values which confirmed complete solubilisation of all triblock copolymers in this solvent. This confirmed that a low amount of acid was needed for the dixanthate PBT and PBT-poly(acrylate) triblock copolymers to be molecularly dissolved in the SEC eluent.
| Polymer | M/mmol | AIBN/mmol | V TCE/mL | t/h | Conv.a | M n(theory) /g mol−1 | M n(SEC) /g mol−1 | Đ | x PBT th | x PBT exp, CDCl3/CHCl2COOH | x PBT exp, CDCl3 |
|---|---|---|---|---|---|---|---|---|---|---|---|
|
a Conversion determined by 1H NMR (CDCl3).
b
M
n (theory) = ([M]0/[PBT]0) × conv × MW(M) + Mn,NMR PBT.
c
M
n and dispersities Đ determined by RI-SEC in a chloroform : dichloroacetic acid mixture (93:7 vol.%) calibration by PS standards.
d
M
n and Đ determined by RI-SEC in chloroform calibration by PS standards.
e
x
PBT = wPBT/(wPBT + 220(1 − wPBT))/MW(M)). wPBT = mPBT/(mPBT + conv × mM).
f Determined by 1H NMR (CDCl3).
g Determined by 1H NMR (CDCl3 : CHCl2COOH).
|
|||||||||||
| PBTA Macroinitiator | 3500 | 1.32 | |||||||||
| PBTB Macroinitiator | 3150 | 1.36 | |||||||||
| P(n-BA)-b-PBTA-b-P(n-BA) 1 | 3.9 | 0.018 | 2.5 | 24 | 0.99 | 3350 | 4900 | 1.48 | |||
| P(n-BA)-b-PBTA-b-P(n-BA) 2 | 4.6 | 0.024 | 1 | 24 | 0.99 | 8400 | 9800 | 1.92 | 0.20 | 0.18 | 0.13 |
| P(n-BA)-b-PBTA-b-P(n-BA) 3 | 3.9 | 0.018 | 0.5 | 24 | 0.99 | 12000 | 14450 |
2.51 | 0.13 | 0.13 | 0.10 |
| P(n-BA)-b-PBTB-b-P(n-BA) 1 | 4.1 | 0.018 | 1.5 | 24 | 0.99 | 6150 | 5350 | 1.53 | |||
| P(t-BA)-b-PBTA-b-P(t-BA) 1 | 3.9 | 0.024 | 1 | 24 | 0.93 | 7150 | 8300 | 1.66 | 0.24 | 0.22 | 0.15 |
| P(t-BA)-b-PBTA-b-P(t-BA) 2 | 3.9 | 0.024 | 0.5 | 24 | 0.95 | 11950 |
12400 |
2.07 | 0.13 | 0.11 | 0.07 |
| P(t-BA)-b-PBTB-b-P(t-BA) 1 | 2.1 | 0.018 | 2 | 24 | 0.99 | 4300 | 4150 | 1.48 | |||
| P(t-BA)-b-PBTB-b-P(t-BA) 2 | 4.0 | 0.036 | 2 | 24 | 0.99 | 6100 | 5600 | 1.50 | |||
| P(t-BA)-b-PBTB-b-P(t-BA) 3 | 4.0 | 0.036 | 1 | 4 | 0.88 | 7400 | 6450 | 1.72 | |||
| P(t-BAm)-b-PBTA-b-P(t-BAm) 1 | 4.7 | 0.021 | 2.5 | 6 | 0.99 | 5000 | 9650 (5500)d | 3.55 (1.78)d | 0.37 | 0.34 | 0.28 |
| P(t-BAm)-b-PBTA-b-P(t-BAm) 2 | 4.1 | 0.018 | 2 | 6 | 0.99 | 6650 | 12650 (6150)d |
5.26 (1.88)d | 0.26 | 0.24 | 0.22 |
| P(t-BAm)-b-PBTA-b-P(t-BAm) 3 | 4.1 | 0.018 | 2 | 6 | 0.99 | 11850 |
26700 |
7.20 | 0.13 | 0.10 | 0.08 |
The Mn of the resulting triblock copolymers of P(t-BA) and P(n-BA) from both PBTA and PBTB at high conversions were in good agreement with theoretical estimations for a controlled polymerization (Table 2).
The monomodal SEC chromatograms (Fig. 3) of the triblock copolymers were shifted to the higher molar mass region from the PBT macroinitiators. These results showed that the two PBT RAFT/MADIX agents coming from both method A and B were able to activate and control the polymerization of n-BA and t-BA. Dispersities increased with molar mass of triblock copolymers, i.e. when the initial concentration of PBT precursor was decreased. This may be partly due to an increasing proportion of irreversibly terminated chains with increasing theoretical Mn although the initiator concentration was purposely selected low to minimize this undesired effect. A more plausible explanation to this broadening of molar mass distributions could be the presence of PBT-P(Acrylate) diblock copolymer in the final product due to the contamination of the dixanthate PBT precursor by a small proportion of monoxanthate PBT (as revealed by MALDI-TOF MS). The triblock copolymer chains should contain twice as much acrylate units as the diblock analogs in the sample, thus leading to a marked influence on Đ values when the diblock fraction is non-negligible and/or at high acrylate content in the final copolymer.
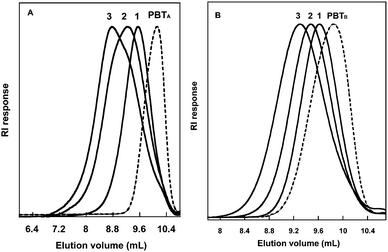 | ||
| Fig. 3 Size exclusion chromatograms (RI response) in chloroform:dichloroacetic acid (93:7 vol.%) of the PBTA macroinitiator (A, dotted line) and triblock copolymers of P(n-BA) (A, solid line) and of the PBTB macroinitiator (B, dotted line) and triblock copolymers of P(t-BA) (B, solid line). | ||
The SEC chromatograms of triblock copolymers of P(t-BAm) were also shifted to the high molar mass region from the PBTA macroinitiator (Fig. 4-A). However, an unexpected tailing was observed in the high molecular weight region of the triblock copolymers when the mixture chloroform : dichloroacetic acid (93:7 vol.%) was used as the eluent (Fig. 4-A). This unexpected phenomenon may be explained by a partial aggregation of copolymer chains induced by the presence of acid in the SEC eluent. This point is currently under investigation in our group. Consequently, Mn values determined with a PS calibration were nearly twice higher than expected (see Table 2) and Đ increased up to abnormally high values (Đ = 7.2 for last entry of Table 2) with increasing targeted Mn. In contrast, the observed tailing disappeared in pure chloroform (Fig. 4-B). In the latter case, the Mn and Đ values were similar to those of polyacrylate-based triblock copolymers determined in the chloroform : dichloroacetic acid eluent phase, for identical theoretical Mn. We explained this result by the absence of polymer aggregates when no dichloroacetic acid was present in the mobile phase, with relatively good elution characteristics in spite of the partial solubilisation of the PBT mid-block in chloroform as previously observed by 1H NMR.
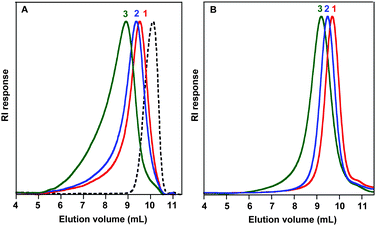 | ||
| Fig. 4 Size exclusion chromatograms (RI response) of the PBTA macroinitiator (dotted line) and triblock copolymers of P(t-BAm) (solid line) in chloroform : dichloroacetic acid (93:7 vol.%) (A) and in pure chloroform (B). | ||
A RAFT/MADIX polymerization of t-BA in the presence of PBTB was followed over time in order to estimate the controlled nature of the polymerization. We followed the evolution profile of macromolecular characteristics during the polymerization. As shown in Fig. 5-b, the SEC chromatograms of the triblock copolymers were shifted to the higher molar mass region when the conversion increased. The evolution of the molar masses with conversion of t-BA was close to the theoretical curve (Fig. 5-a) with slightly higher Mn than expected in the early stages of the polymerization. Moreover, Đ values were relatively high for a RDRP (1.50 < Đ < 1.72) and remained relatively unchanged as the reaction proceeded. This evolution of Mn and Đ during polymerization is typical of that previously observed for the RAFT/MADIX polymerization of acrylates mediated by O-ethyl xanthates.55,56 Recently, a transfer constant CX to Rhodixan A1 equal to 1.66 ± 0.38 and an interchain transfer constant Cex equal to 2.5 ± 0.2 were determined for the bulk polymerization of t-BA at 60 °C.57 This means that both transfer to xanthate and interchain transfer are relatively slow compared to propagation, and explain the profiles observed for Mn and Đ.58 The structure of Rhodixan A1 (CH3CH(CO2CH3)S(CS)OCH2CH3) represents an excellent model for the xanthate end groups of PBT whose reactivity is thought to be in the same order of magnitude. It is however expected that the dixanthate PBT precursor should react roughly twice as fast than Rhodixan A1 because it contains twice its number of transferring sites. These results revealed that the dixanthate-terminated PBT was an appropriate RAFT/MADIX agent for synthesis of well-controlled P(t-BA)-PBT-P(t-BA) triblock copolymers.
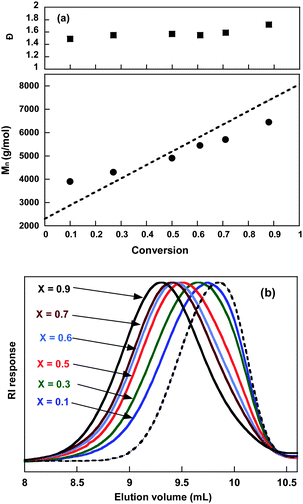 | ||
| Fig. 5 RAFT/MADIX polymerization of t-BA from the PBTB macroinitiator. T = 70 °C. (a) Evolution of Mn and Đ with t-BA conversion. (b) Size exclusion chromatograms (RI response) in chloroform : dichloroacetic acid (93:7 vol.%) of the PBTB macroinitiator (dotted line) and triblock copolymers of P(t-BA) (solid line). | ||
Conclusions
Well-defined ABA triblock copolymers comprising a PBT central block were synthesized by combining step-growth and RAFT/MADIX polymerizations. Acrylate and acrylamido monomers were selected to prove the feasibility of this approach. Prior to RAFT/MADIX polymerization, various low molar mass α,ω-dixanthate PBTs were synthesized via two distinct synthetic routes. In the first approach, 1H NMR and MALDI-TOF MS analysis confirmed that the transformation of terminal hydroxyl functionalities of PBT into O-ethyl xanthate end-groups was quantitative. The PBT macro-RAFT/MADIX agent was also successfully prepared through the direct use of a xanthate chain stopper in the step-growth polymerization. In the latter case, DPn was controlled by varying the concentration of the hydroxyxanthate chain stopper. These first results pave the way to many synthetic possibilities considering the unique choice of monomer combinations offered by the RAFT/MADIX process mediated by O-ethyl xanthates.Acknowledgements
The authors are grateful to Rhodia and CNRS for the financial support of this work.References
- M. A. Tasdelen and Y. Yagci, Macromol. Eng., 2007, 1, 541–604 Search PubMed.
- Y. Yagci and M. A. Tasdelen, Prog. Polym. Sci., 2006, 31, 1133–1170 CrossRef CAS.
- A. D. Jenkins, R. G. Jones and G. Moad, Pure Appl. Chem., 2010, 82, 483–491 CrossRef CAS.
- M. H. Acar and K. Matyjaszewski, Macromol. Chem. Phys., 1999, 200, 1094–1100 CrossRef CAS.
- A. M. Morgan, S. K. Pollack and K. Beshah, Macromolecules, 2002, 35, 4238–4246 CrossRef CAS.
- M. Nasser-Eddine, C. Delaite, G. Hurtrez and P. Dumas, Eur. Polym. J., 2005, 41, 313–318 CrossRef CAS.
- L. C. Gao, C. L. Zhang, X. Liu, X. H. Fan, Y. X. Wu, X. F. Chen, Z. Shen and Q. F. Zhou, Soft Matter, 2008, 4, 1230–1236 RSC.
- M. K. Mahanthappa, F. S. Bates and M. A. Hillmyer, Macromolecules, 2005, 38, 7890–7894 CrossRef CAS.
- T. Masukawa, A. Yokoyama and T. Yokozawa, Macromol. Rapid Commun., 2009, 30, 1413–1418 CrossRef CAS.
- C.-F. Huang, A. Yokoyama and T. Yokozawa, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2948–2954 CrossRef CAS.
- Y. J. Kim, M. Seo and S. Y. Kim, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1049–1057 CrossRef CAS.
- Y. Yamazaki, N. Ajioka, A. Yokoyama and T. Yokozawa, Macromolecules, 2009, 42, 606–611 CrossRef CAS.
- J. Liu, E. Sheina, T. Kowalewski and R. D. McCullough, Angew. Chem., Int. Ed., 2002, 41, 329–332 CrossRef CAS.
- K. Y. Choi and K. B. McAuley, Polym. React. Eng., 2007, 273–314 Search PubMed.
- S. G. Gaynor and K. Matyjaszewski, Macromolecules, 1997, 30, 4241–4243 CrossRef CAS.
- S. Zhu, G. Xiao and D. Yan, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2943–2950 CrossRef CAS.
- Y. Zhang, I. S. Chung, J. Huang, K. Matyjaszewski and T. Pakula, Macromol. Chem. Phys., 2005, 206, 33–42 CrossRef CAS.
- K. Tharanikkarasu, H. Verma, W. Jang, S. K. Lee, J. Seo, S. Baek and H. Han, J. Appl. Polym. Sci., 2008, 108, 1538–1544 CrossRef CAS.
- H. Verma and K. Tharanikkarasu, Polym. Int., 2008, 57, 226–232 CrossRef CAS.
- K. Matyjaszewski, Y. Nakagawa and S. G. Gaynor, Macromol. Rapid Commun., 1997, 18, 1057–1066 CrossRef.
- F. J. Xu, J. P. Zhao, E. T. Kang, K. G. Neoh and J. Li, Langmuir, 2007, 23, 8585–8592 CrossRef CAS.
- B. Yameen, M. Alvarez, O. Azzaroni, U. Jonas and W. Knoll, Langmuir, 2009, 25, 6214–6220 CrossRef CAS.
- D. Shen, Y. Shi, Z. Fu, W. Yang, X. Cai, R. Lin and D. Zhang, Polym. Bull., 2003, 49, 321–328 CrossRef CAS.
- M. Mennicken, R. Nagelsdiek, H. Keul and H. Hoecker, Macromol. Chem. Phys., 2004, 205, 143–153 CrossRef CAS.
- K. Sha, D. Li, Y. Li, P. Ai, X. Liu, W. Wang, Y. Xu, S. Wang, M. Wu, B. Zhang and J. Wang, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 3393–3399 CrossRef CAS.
- G. Colomines, B. Otazaghine, C. Boyer, S. Monge and J.-J. Robin, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 433–443 CrossRef CAS.
- E. Yoshida and M. Nakamura, Polym. J., 1998, 30, 915–920 CrossRef CAS.
- S. G. Gaynor, S. Z. Edelman and K. Matyjaszewski, Polym. Prepr., 1997, 38, 703–704 CAS.
- D. Li, K. Sha, Y. Li, X. Liu, W. Wang, S. Wang, Y. Xu, P. Ai, M. Wu and J. Wang, Polym. Bull., 2005, 56, 111–117.
- M. R. Mauricio, G. M. Carvalho, E. Radovanovic, E. C. Muniz and A. F. Rubira, Mater. Sci. Eng., C, 2009, 29, 594–598 CrossRef CAS.
- Handbook of RAFT Polymerization, ed. C. Barner-Kowollik, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed.
- D. Taton, M. Destarac and S. Z. Zard, in Handbook of RAFT Polymerization, ed. C. Barner-Kowollik, Wiley-VCH, Weinheim, Germany, 2008, pp. 373–421 Search PubMed.
- A. B. Lowe and C. L. McCormick, Prog. Polym. Sci., 2007, 32, 283–351 CrossRef CAS.
- D. Taton, A.-Z. Wilczewska and M. Destarac, Macromol. Rapid Commun., 2001, 22, 1497–1503 CrossRef CAS.
- D. Charmot, P. Corpart, H. Adam, S. Z. Zard, T. Biadatti and G. Bouhadir, Macromol. Symp., 2000, 150, 23–32 CrossRef CAS.
- M. Beija, J.-D. Marty and M. Destarac, Chem. Commun., 2011, 47, 2826–2828 RSC.
- M. Destarac, A. Guinaudeau, R. Geagea, S. Mazieres, E. Van Gramberen, C. Boutin, S. Chadel and J. Wilson, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5163–5171 CrossRef CAS.
- M. Destarac and B. Bavouzet, Rhodia Chimie, WO 07/2889194, 2007.
- M. Destarac, G. Mignani, S. Zard, B. Sire and C. Kalai, Rhodia Chimie, WO 02/2812296, 2002.
- G. Pound, F. Aguesse, J. B. McLeary, R. F. M. Lange and B. Klumperman, Macromolecules, 2007, 40, 8861–8871 CrossRef CAS.
- A. K. Mishra, V. K. Patel, N. K. Vishwakarma, C. S. Biswas, M. Raula, A. Misra, T. K. Mandal and B. Ray, Macromolecules, 2011, 44, 2465–2473 CrossRef CAS.
- M. Destarac, W. Bzducha and S. Z. Zard, Polym. Prepr., 2005, 46, 387–388 CAS.
- R. Gallucci and B. R. Patel, in Modern Polyesters, Scheirs, J., Long, T, (ed.), John Wiley, Chichester, UK, 2003, pp. 293–321 Search PubMed.
- P. N. Vashi, A. K. Kulshreshtha and K. P. Dhake, J. Appl. Polym. Sci., 2003, 87, 1190–1193 CrossRef CAS.
- According to the recent recommendations of IUPAC ( R. F. T. Stepto, Pure Appl. Chem., 2009, 81, 351–353 Search PubMed and R. F. T. Stepto, Pure Appl. Chem., 2009, 81, 779 CrossRef ), the term dispersity (Đ = Mw/Mn) is used instead of the misleading term polydispersity.
- B. Boutevin, M. Khamlichi, J. M. Lusinchi and J. J. Robin, Macromol. Chem. Phys., 1997, 198, 605–617 CAS.
- R. Nicolay, Y. Kwak and K. Matyjaszewski, Chem. Commun., 2008, 5336–5338 RSC.
- http://www.shodex.net/ .
- R. Alicata, G. Montaudo, C. Puglisi and F. Samperi, Rapid Commun. Mass Spectrom., 2002, 16, 248–260 CrossRef CAS.
- C. Puglisi, F. Samperi, R. Alicata and G. Montaudo, Macromolecules, 2002, 35, 3000–3007 CrossRef CAS.
- S. Carroccio, P. Rizzarelli, G. Scaltro and C. Puglisi, Polymer, 2008, 49, 3371–3381 CrossRef CAS.
- T. Gruendling, S. Weidner, J. Falkenhagen and C. Barner-Kowollik, Polym. Chem., 2010, 1, 599–617 RSC.
- I. Q. Li, D. M. Knauss, Y. Gong, B. A. Howell and D. B. Priddy, Polym. Prepr., 1999, 40, 383–384 CAS.
- W. H. Carothers, J. Am. Chem. Soc., 1929, 51, 2548–2559 CrossRef CAS.
- M. Destarac, W. Bzducha, D. Taton, I. Gauthier-Gillaizeau and S. Z. Zard, Macromol. Rapid Commun., 2002, 23, 1049–1054 CrossRef CAS.
- P. Chapon, C. Mignaud, G. Lizarraga and M. Destarac, Macromol. Rapid Commun., 2003, 24, 87–91 CrossRef CAS.
- A. Guinaudeau, PhD thesis, University of Toulouse, 2010.
- M. Destarac, Polym. Rev., 2011, 51, 163–187 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary characterization of compounds (1H and 13C NMR spectroscopy and MALDI-TOF MS), supplementary experimental studies. See DOI: 10.1039/c1py00270h |
| This journal is © The Royal Society of Chemistry 2011 |