DOI:
10.1039/C1PY00269D
(Paper)
Polym. Chem., 2011,
2, 2557-2563
Received
13th June 2011
, Accepted 5th August 2011
First published on 1st September 2011
Abstract
Thioxanthone-ethylcarbazole (TX-EC), 7-ethylthiochromeno[2,3-b]carbazol-13(7H)-one, as a novel visible light absorbing one-component Type II photoinitiator with high solubility, was synthesized and characterized. Its ability to initiate free radical photopolymerization of methyl methacrylate (MMA) was demonstrated and compared with that of the parent thioxanthone-carbazole (TX-C) both in the presence and absence of a co-initiator. It is found that TX-EC is a more effective photoinitiator in all cases. The mechanism of initiating radical formation for free radical polymerization in the absence of an added co-initiator involves photoexcitation of TX-EC and hydrogen abstraction of triplet states of TX-EC from the ethyl group attached to the carbazole moiety. In the presence of triethyl amine, the corresponding hydrogen abstraction reaction occurs between a triplet photoinitiator and the amine compound. The possibility of visible light photoinitiation of free radical promoted cationic polymerization of several monomers, namely cyclohexene oxide (CHO), n-butyl vinylether (BVE), N-vinyl carbazole (NVC) and 3,4-epoxycyclohexyl-3′,4′-epoxycyclohexene carboxylate (EEC) using TX-EC and Ph2I+PF6− combination was also demonstrated.
Introduction
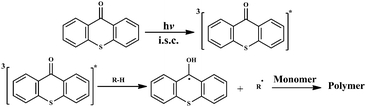
Among various Type II photoinitiators, thioxanthone (TX) and its derivatives are the most widely used compounds in various UV curing applications because of their excellent light absorption characteristics in the near UV region.1 In general, the photoinitiating free radicals are generated by the hydrogen abstraction of triplet excited states of TX from hydrogen donors such as amines, alcohols, ethers and thiols as depicted in Scheme 1.2–4
As with all the other Type II photoinitiators, TXs and their derivatives suffer from several distinct disadvantages including requirement of hydrogen donors, low solubility and low sensitivity in the visible region. Low molecular-weight amines are the most widely used hydrogen donors for TX based systems in many UV curing applications. However, particularly when used at high concentrations, they have odor, toxicity5 and migration problems and cause a decrease in the pendulum hardness of the cured films.6 The commonly applied approach to overcome these problems is to chemically incorporate the hydrogen donating sites into the TX chromophores.7–9 We have previously reported several thiol10 and acetic acid11 derivatives of TX as photoinitiators for free radical polymerization. A major advantage of this type of initiators is related to their one component nature.12 They can serve as both a triplet photosensitizer and a hydrogen donor. This way, the odor and toxicity problems observed with the conventional photoinitiators and amine hydrogen donors were overcome. We have also reported several polymeric benzophenone13 and thioxanthone14,15 photoinitiators that avoid migration problems. More recently, we have succeeded in extending the optical absorption properties of TXs to near-UV and visible regions by incorporating polynuclear aromatic moieties such as anthracene,16 fluorene17,18 and carbazole18,19 chromophoric groups into the TX structure. However, except in dimethyl formamide most of these photoinitiators were not soluble in monomers and common organic solvents limiting their practical use in UV curing applications.
As part of our major focus in developing new photoinitiating systems to overcome the existing problems or enhance the efficiency of UV-curable formulations, herein we report the synthesis, characterization, and photoinitiation capability of thioxanthone-ethylcarbazole (TX-EC). In addition to excellent absorption characteristics in the visible range, this photoinitiator exhibits high solubility in common polar and nonpolar solvents, and monomers. Moreover, due to the presence of hydrogen donating sites adjacent to the N atom of the carbazole group, TX-EC displays one-component nature and can act as an efficient photoinitiator in the visible light region for free radical and free radical promoted cationic photopolymerization without the necessity for an additional hydrogen donor.
Experimental part
Materials
Thiosalicylic acid (97%, Sigma-Aldrich), ethyl-carbazole (EC, 99%, Merck) and diphenyliodonium hexafluorophosphate were used without further purification. Cyclohexene oxide (CHO, 98%, Aldrich), n-butyl vinylether (BVE, >97%, Fluka) and triethylamine (TEA, >99.5%, Fluka) were distilled over CaH2 before use in vacuo. N-Vinyl carbazole (NVC, 98%, Aldrich) was crystallized from ethanol. 3,4-Epoxycyclohexyl-3′,4′-epoxycyclohexene carboxylate (EEC), a commercial product of Ciba Specialty Chemicals, CY-179, was used as received. Methyl methacrylate (MMA, 99%, Aldrich) was passed through a basic alumina column before use to remove the inhibitor. Trimethylolpropane triacrylate (TMPTA, 95%, Aldrich) was used as received. p-Dioxane (99.9%, JT Baker), sulfuric acid (H2SO4, 95–97%, Fluka), and ammonium hydroxide (0.1 N in H2O, Aldrich) were used as received. All other solvents were purified by conventional procedures.
Thiosalicylic acid (0.2 g, 1.3 mmol) was slowly added to concentrated sulfuric acid (10 mL), and the mixture was stirred for 5 min to ensure thorough mixing. EC (0.8 g, 3.9 mmol) was added slowly to the stirred mixture over a period of 30 min. After the addition, the reaction mixture was stirred at 75 °C for 2 hours and later it was left to stand at room temperature overnight. Afterwards, the resulting mixture was poured carefully with stirring into a 10-fold excess of boiling water, and it was then boiled further for 5 min. The solution was cooled, filtered and washed further with aqueous ammonia. The residue was recrystallized from dioxane/water mixture to give a pure bright yellow solid. Yield: 53%; 1H NMR (CDCI3, 250 MHz): δ = 9.3–7.4 (m, 10H, aromatic), 4.5 (q, 2H, CH), 1.6–2 (m, 3H, CH), FT-IR (ATR): 3100, 3000, 1335, 1250, 1662 cm−1.
Appropriate solutions of the monomer and TX-EC were irradiated with a Polilight PL400 Forensic Plus light source at 415 nm in a nitrogen atmosphere. Polymers were obtained after precipitation in methanol and drying under vacuum. Conversions were calculated gravimetrically for all samples.
Instrumentation
1H NMR spectra of 5–10% (w/w) solutions in CDCl3 with Si(CH3)4 as an internal standard were recorded at room temperature at 250 MHz on a Bruker DPX 250 spectrometer. FT-IR spectra were recorded on a Perkin-Elmer FTIR Spectrum One spectrometer via the attenuated total reflectance (ATR) technique with 4 scans for each sample. UV-visible spectra were recorded on a Shimadzu UV-1601 UV-visible spectrophotometer. Differential scanning calorimeter (DSC) was performed on a Perkin Elmer Diamond DSC with a heating rate of 10 °C min−1 under nitrogen flow. Gel permeation chromatography (GPC) measurements were obtained from a Viscotek GPCmax Autosampler system consisting of a pump, a Viscotek UV detector and a Viscotek differential refractive index (RI) detector. Three ViscoGEL GPC columns (G2000HHR, G3000HHR and G4000HHR, 7.8 mm internal diameter, 300 mm length) were used in series. The effective molecular weight ranges were 456–42![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800, 1050–107000, and 10200–2890000, respectively. THF was used as an eluent at a flow rate of 1.0 mL min−1 at 30 °C. Both detectors were calibrated with polystyrene standards having a narrow molecular weight distribution. Data were analyzed using Viscotek OmniSEC Omni-01 software. Molecular weights were calculated with the aid of polystyrene standards. Fluorescence measurement was performed on a Jobin Yvon-Horiba Fluoromax-P spectrophotometer. A Rofin Polilight PL400 Forensic Plus light source, emitting light at several wavelengths (350, 415, 430, 450, LP530, 490, 505, 515, 530, 550, 560, LP560, 570, 590, 620, 650, white, half white and blank) with specific bandwidths, was used for the irradiation of the samples.
800, 1050–107000, and 10200–2890000, respectively. THF was used as an eluent at a flow rate of 1.0 mL min−1 at 30 °C. Both detectors were calibrated with polystyrene standards having a narrow molecular weight distribution. Data were analyzed using Viscotek OmniSEC Omni-01 software. Molecular weights were calculated with the aid of polystyrene standards. Fluorescence measurement was performed on a Jobin Yvon-Horiba Fluoromax-P spectrophotometer. A Rofin Polilight PL400 Forensic Plus light source, emitting light at several wavelengths (350, 415, 430, 450, LP530, 490, 505, 515, 530, 550, 560, LP560, 570, 590, 620, 650, white, half white and blank) with specific bandwidths, was used for the irradiation of the samples.
Photocalorimetry (photo-DSC)
The photo-differential scanning calorimetry (photo-DSC) measurements were carried out by means of a modified Perkin-Elmer Diamond DSC equipped with a high pressure mercury arc lamp (320–500 nm). A uniform UV light intensity is delivered across the DSC cell to the sample and reference pans. The intensity of the light was measured as 53 mW cm−2 by a UV radiometer capable of broad UV range coverage. The mass of the sample was 3 mg, and the measurements were carried out in an isothermal mode at 30 °C under a nitrogen flow of 20 mL min−1. The reaction heat liberated in the polymerization was directly proportional to the number of acrylate groups reacted in the system. By integrating the area under the exothermic peak, the conversion of the acrylate groups (C) or the extent of the reaction was determined according to eqn (1):where ΔHt is the reaction heat evolved at time t and ΔH0theory is the theoretical heat for complete conversion. ΔH0theory = 86 kJ mol−1 for an acrylic double bond.20 The rate of polymerization (Rp) is directly related to the heat flow (dH/dt) by eqn (2):| | | Rp = dC/dt = (dH/dt)/ΔH0theory | (2) |
Results and discussion
Synthesis and characterization of the photoinitiator
To investigate the effect of the alkyl group on the apparent state of TX-C photoinitiators, the TX photoinitiator with an ethyl carbazole group was synthesized and characterized. Thus, TX-EC was successfully prepared from thiosalicylic acid by conventional thioxanthone ring forming reaction21 using ethylcarbazole (EC) (Scheme 2).
 |
| | Scheme 2 Synthesis of TX-EC. | |
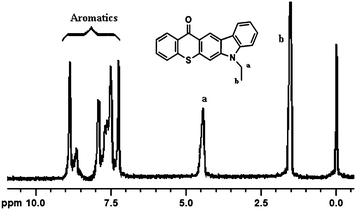
The structure of the photoinitiator was confirmed by 1H NMR, UV, and fluorescence spectral analysis. As can be seen from Fig. 1, the 1H NMR spectrum of the photoinitiator exhibits not only the specific signals of the thioxanthone ring but also chemical shifts belonging to the alkyl chain and carbazole. The peaks characteristic of aromatic protons of thioxanthone and carbazole appeared between 9.3 and 7.4 ppm. Furthermore, the two new signals corresponding to ethyl (N–CH2CH3) protons were detectable at 4.5 and 1.6–2 ppm. On the basis of a comparison of the integration of the peak intensities at 9.3–7.4 and 4.5 ppm, respectively, the obtained ratio of 5:1 clearly indicates the efficiency of the reaction.
Introducing the ethyl group to TX-C drastically changes the solubility behavior of the bare photoinitiator. As can be seen from Table 1, TX-EC is soluble in highly polar solvents such as DMF as well as in the less polar solvents such as THF and n-hexane.
Table 1 Solubility of TX-C and TX-EC in polar and nonpolar solvents
Absorption and emission characteristics of the obtained thioxanthone compound were investigated by UV and fluorescence spectroscopy. Fig. 2 demonstrates the comparison of the UV spectra of TX-EC with the parent compounds, TX and EC. As can be seen, the new substituted photoinitiator exhibits absorption characteristics very similar to TX, except a tail absorption in the visible wavelength region (λ > 400 nm), where the TX chromophore is transparent. Although EC itself does not absorb the light at above 350 nm, it has a dramatic effect on the absorption characteristics particularly at high wavelengths due to the extended conjugation. Fluorescence spectra of the photoinitiator may also provide information on the nature of the excited states involved.
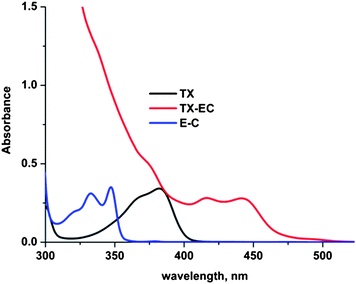 |
| | Fig. 2 UV spectra of TX-EC, TX, and EC at equal concentrations (1.0 × 10−5 M) in CH2Cl2. | |
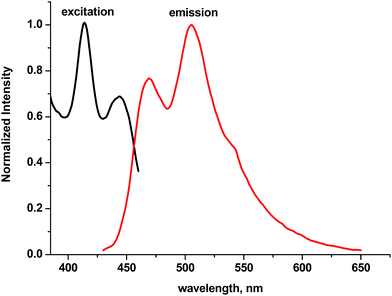
As can be seen from Fig. 3, excitation and emission fluorescence spectra in CH2Cl2 of TX-EC shows a nearly mirror-image-like relation between absorption and emission again similar to bare TX, indicating its dominant photoexcited (singlet) state in the photoinitiator.
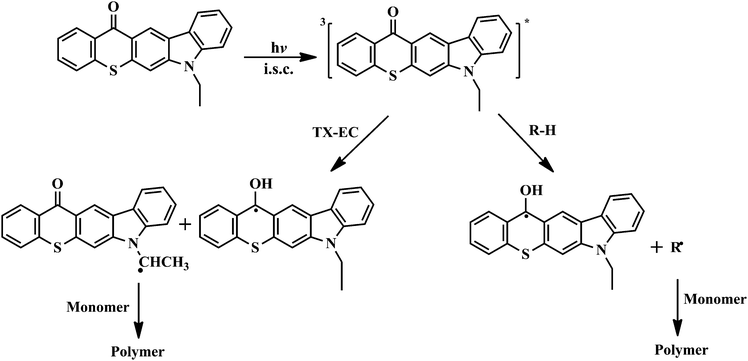
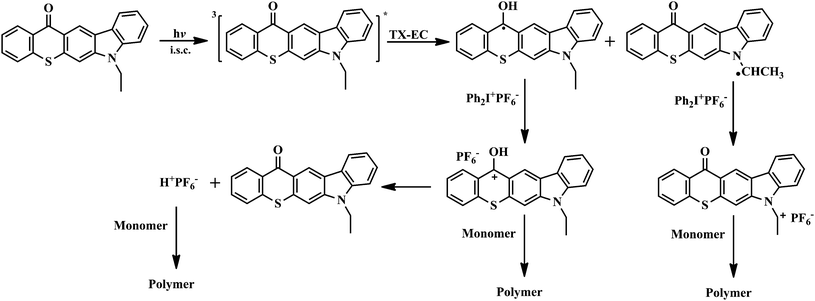
As demonstrated for previously reported highly conjugated TX derivatives,16–19 TX-EC is expected to undergo an irreversible photolysis in the presence and absence of hydrogen donors. This was further confirmed for TX-EC by the spectral changes on UV irradiation. Representative results for the photobleachings of TX-EC in both cases are shown in Fig. 4. UV spectra were recorded after the solution had been exposed to the visible light for subsequent intervals. TX-EC was found to be an efficient initiator for visible light photopolymerization in the presence and absence of a hydrogen donor in accordance with the photobleaching results. The proposed mechanism considering both cases is presented in Scheme 3.
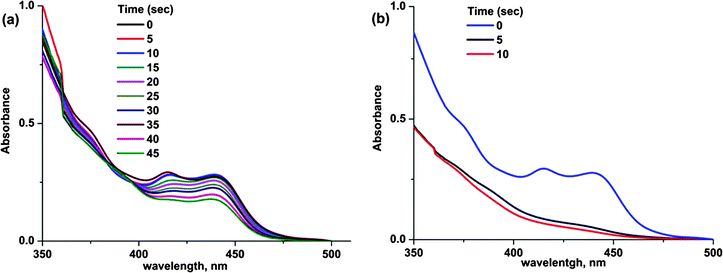 |
| | Fig. 4 Photobleaching of TX-EC (1.0 × 10−5 M) in CH2Cl2 in the presence (a) and absence (b) of TEA (1.0 × 10−4 M). | |
Table 2 Photoinitiated polymerizationa of MMA using, TX-EC at 415 nm in bulk
| TEA/mol L−1 |
N2 |
Conversion (%) |
M
n
/g mol−1 |
M
w/Mn |
|
TEA, triethylamine, irradiation time = 15 min. [MMA] = 9.38 mol L−1, [TX-EC] = 5 × 10−3 mol L−1.
Determined by chromatography (GPC) using polystyrene standards.
|
| 5 × 10−2 |
− |
9.6 |
8800 |
2.20 |
| 5 × 10−2 |
+ |
12.8 |
9770 |
2.23 |
| — |
+ |
4.0 |
63,350 |
2.29 |
As expected, TX-EC was found to efficiently initiate photopolymerization at this region even in the absence of an additional hydrogen donor. Notably, the addition of a hydrogen donor such as triethylamine (TEA) accelerates the polymerization. The efficiency of the TX-EC in the photocuring of formulations containing multifunctional monomers was also studied. The heat released during the photocuring of the formulations was followed by photo-DSC. In Fig. 5 and 6, kinetic profiles referring to the polymerization of trimethylolpropane triacrylate (TMPTA) under polychromatic light emitting at λ = 315–450 nm are shown. TX-EC and TEA served as the photoinitiator and hydrogen donor, respectively. The shape of the curves indicates the existence of two stages: a rapid first stage followed by a slow stage. It can also be seen that polymerization takes place more rapidly with the hydrogen donor.
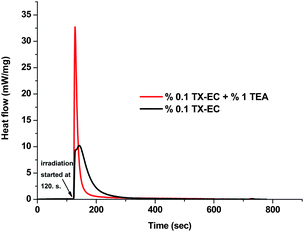 |
| | Fig. 5 Photo-DSC profiles for the photopolymerization of TMPTA irradiated at 30 °C by UV light with an intensity of 58 mW cm−2. | |
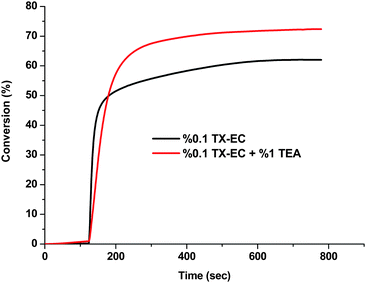 |
| | Fig. 6 Conversion vs. time curves for the photopolymerization of TMPTA irradiated at 30 °C by UV light with an intensity of 58 mW cm−2. | |
In the present study, the free radical promoted cationic photopolymerizations were performed under irradiation at 415 nm at the wavelength where the light emission is well matched with the absorption of TX-EC and the iodonium salt is transparent. Cyclohexene oxide (CHO) was selected as the cationically polymerizable monomer as it structurally represents epoxides, which are used in acid-sensitive, positive-tone, chemically amplified resists, designed for 3D microfabrication and readily undergoes polymerization with cationic species and is inert to free radicals.
Apart from CHO, vinyl compounds such as n-butyl vinyl ether (BVE) and N-vinyl carbazole (NVC), and epoxy resins such as 3,4-epoxycyclohexyl-3′,4′-epoxycyclohexene carboxylate (EEC) were also studied in the photopolymerization reactions. As can be seen from Table 3, all photopolymerizations were conducted quite effectively without hydrogen donors and within a short period of irradiation, 15 minutes. In the case of IBVE and NVC, fast polymerizations were induced even in the absence of hydrogen donors. For these monomers oxidizable radicals are also produced by radical addition to the monomer as demonstrated18 for the other TX derivatives in the example of BVE where Ph2I+PF6− is the oxidant. Because of the presence of two epoxide groups, EEC readily crosslinks and ultimately forms gelled systems.
Table 3 Radical promoted cationic photopolymerization of various monomersa using TX-EC, in conjunction with Ph2I+PF6− (λ = 415 nm) in CH2Cl2
| Monomers |
Conversionb (%) |
M
n
|
M
w/Mnc |
|
[Monomer] = 0.97 mol L−1, [TX-EC]= 4.8 × 10−3 mol L−1, [Ph2I+PF6−] = 4.8 × 10−3 mol L−1.
Determined gravimetrically after precipitation in methanol.
Determined by GPC using polystyrene standards.
EEC is used as a multifunctional monomer, gelation time = 13 min.
|
| CHO |
72.6 |
9790 |
1.82 |
| IBVE |
80.6 |
15,900 |
1.55 |
| NVC |
95.4 |
818,100 |
1.46 |
| EECd |
99 |
∞ |
— |
In order to define the actual initiating species for both free radical and free radical promoted cationic polymerization, we have investigated the UV absorption spectra of the linear polymers thus formed. In these investigations, the chromophore concentrations were the same as adjusted by using the molar extinction coefficient of TX-EC and the molecular weight of the polymers. As stated above, in the free radical systems, although at a lower rate, TX-EC displays photoinitiation activity even in the absence of a hydrogen donor.
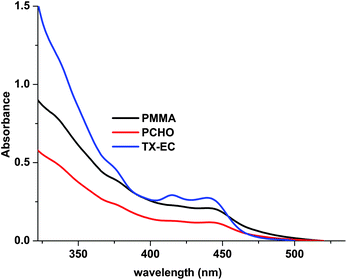
In this case, triplet TX-EC reacts with ground-state TX-EC by hydrogen abstraction to produce ethyl radicals which initiate the polymerization, and gets incorporated into the polymer (TX-EC polymer). The TX-EC moiety, which is covalently attached to the polymer, is clearly shown by UV-vis spectroscopy. As can be seen from Fig. 7, the UV-vis spectra of TX-EC and the corresponding polymer (PMMA) in CH2Cl2 solutions are quite similar. A comparable analysis was also conducted for the cationic polymerization. The UV spectrum of the corresponding polymer, PCHO exhibits relatively lower absorption at the visible range. The observed absorbance differences can be explained in terms of initiation mechanisms. As depicted in Scheme 4, besides the oxidation of amino alkyl radicals which leads to the formation of polymer chains with TX-EC moieties, the initiation through protonic acids may also be operative. In the latter case, the oxidation of TX ketyl radicals ultimately releases protonic acids capable of initiating cationic polymerization. The polymer chains formed this way do not contain any chromophoric TX-EC groups. The initiation mechanisms considering both pathways are depicted in Scheme 4.
Conclusions
In this work, we successfully synthesized a new photoinitiator possessing additional chromophoric and alkyl groups facilitating extended absorption in the visible range and solubility, respectively. This novel photoinitiator initiates the free radical polymerization in the absence and presence of a hydrogen donor. In addition, TX-EC in conjunction with an iodonium salt promotes the cationic polymerization of various industrially important monomers in the visible range without the requirement of any hydrogen donor. Initiation of both cationic and radical polymerization suggests that this photoinitiator can be used for practical applications involving hybrid monomers based on epoxides and (meth)acrylates.
Acknowledgements
The authors would like to thank Istanbul Technical University for financial support. One of the authors (Y.Y.) thanks the Turkish Academy of Sciences for partial financial support.
Notes and references
- R. S. Davidson, Adv. Phys. Org. Chem., 1983, 19, 1–130 CrossRef CAS.
- M. V. Encinas, A. M. Rufs, T. Corrales, F. Catalina, C. Peinado, K. Schmith, M. G. Neumann and N. S. Allen, Polymer, 2002, 43, 3909–3913 CrossRef CAS.
- T. Corrales, F. Catalina, C. Peinado, N. S. Allen, A. M. Rufs, C. Bueno and M. V. Encinas, Polymer, 2002, 43, 4591–4597 CrossRef CAS.
- T. Corrales, F. Catalina, N. S. Allen and C. Peinado, J. Photochem. Photobiol., A, 2005, 169, 95–100 CrossRef CAS.
- B. Vazquez, B. Levenfeld and J. S. Roman, Polym. Int., 1998, 46, 241–250 CrossRef CAS.
-
S. Paul, Surface Coating: Science and Technology, Wiley-Interscience, 1986 Search PubMed.
- X. X. Jiang and H. Yin, Polymer, 2004, 45, 133–140 CrossRef CAS.
- X. Y. Jiang, Polymer, 2004, 45, 5057–5063 CrossRef CAS.
- L. Pouliquen, X. Coqueret, F. Morlet-Savary and J.-P. Fouassier, Macromolecules, 1995, 28, 8028–8034 CrossRef CAS.
- L. Cokbaglan, N. Arsu, Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2003, 36, 2649–2653 CrossRef CAS.
- M. Aydin, N. Arsu, Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2005, 38, 4133–4138 CrossRef CAS.
-
M. Aydin, N. Arsu, Y. Yagci, S. Jockusch and N. J. Turro, Photochemistry and UV Curing: New Trends 2006, Research Signpost, 2006 Search PubMed.
- G. Temel, B. Aydogan, N. Arsu and Y. Yagci, Macromolecules, 2009, 42, 6098–6106 CrossRef CAS.
- B. Gacal, H. Akat, D. K. Balta, N. Arsu and Y. Yagci, Macromolecules, 2008, 41, 2401–2405 CrossRef CAS.
- H. Akat, B. Gacal, D. K. Balta, N. Arsu and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2109–2114 CrossRef CAS.
- D. K. Balta, N. Arsu, Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2007, 40, 4138–4141 CrossRef CAS.
- G. Yilmaz, B. Aydogan, G. Temel, N. Arsu, N. Moszner and Y. Yagci, Macromolecules, 2010, 43, 4520–4526 CrossRef CAS.
- G. Yilmaz, S. Beyazit and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1591–1596 CrossRef CAS.
- G. Yilmaz, A. Tuzun and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5120–5125 CrossRef CAS.
- E. Andrzejewska and M. Andrzejewski, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 665–673 CrossRef CAS.
- F. Catalina, J. M. Tercero, C. Peinado, R. Sastre, J. L. Mateo and N. S. Allen, J. Photochem. Photobiol., A, 1989, 50, 249–258 CrossRef CAS.
- Y. Yagci and I. Reetz, Prog. Polym. Sci., 1998, 23, 1485–1538 CrossRef CAS.
- T. Corrales, F. Catalina, C. Peinado and N. S. Allen, J. Photochem. Photobiol., A, 2003, 159, 103–114 CrossRef CAS.
- E. A. Lissi, J. Garrido and A. Zanocco, J. Polym. Sci., Part C: Polym. Lett., 1984, 22, 391–393 CAS.
- A. Bottcher, K. Hasebe, G. Hizal, Y. Yagci, P. Stellberg and W. Schnabel, Polymer, 1991, 32, 2289–2293 CrossRef.
- W. D. Cook, Polymer, 1992, 33, 600–609 CrossRef CAS.
- Y. Yagci, J. Borbely and W. Schnabel, Eur. Polym. J., 1989, 25, 129–131 CrossRef CAS.
- C. Dursun, M. Degirmenci, Y. Yagci, S. Jockusch and N. J. Turro, Polymer, 2003, 44, 7389–7396 CrossRef CAS.
- Y. Yagci, I. Kminek and W. Schnabel, Eur. Polym. J., 1992, 28, 387–390 CrossRef CAS.
- Y. Yagci, I. Kminek and W. Schnabel, Polymer, 1993, 34, 426–428 CrossRef CAS.
- Y. Y. Durmaz, M. Kukut, N. Moszner and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4793–4799 CrossRef CAS.
- J. Lalevee, A. Dirani, M. El-Roz, X. Allonas and J. P. Fouassier, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3042–3047 CrossRef CAS.
- M. El-Roz, J. Lalevee, X. Allonas and J. P. Fouassier, Macromolecules, 2009, 42, 8725–8732 CrossRef CAS.
- J. Lalevee, R. Shankar, M. A. Tehfe, U. Sahoo and J. P. Fouassier, Macromol. Chem. Phys., 2011, 212, 806–812 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.