Thermally reactive Thiazolo[5,4-d]thiazole based copolymers for high photochemical stability in polymer solar cells†
Martin
Helgesen
*,
Morten V.
Madsen
,
Birgitta
Andreasen
,
Thomas
Tromholt
,
Jens W.
Andreasen
and
Frederik C.
Krebs
Risø National Laboratory for Sustainable Energy, Technical University of Denmark, Frederiksborgvej 399, DK-4000, Roskilde, Denmark. E-mail: manp@risoe.dtu.dk
First published on 21st September 2011
Abstract
New thermally reactive copolymers based on dithienylthiazolo[5,4-d]thiazole (DTZ) and silolodithiophene (SDT) have been synthesized and explored in bulk heterojunction solar cells as mixtures with [6,6]-phenyl-C61-butyric acid methyl ester (PCBM). In thin films the polymers had optical band gaps in the range of 1.64–1.80 eV. For solubility the polymers have incorporated alkyl groups on the SDT unit and thermally removable ester groups on the DTZ unit that can be eliminated around 200 °C for improved photochemical stability in thin films. The bulkiness of the alkyl chains on the SDT unit proved to be very significant in terms of photovoltaic performance of the polymer:PCBM solar cells. Polymers based on 4,4-dihexyl-4H-silolo[3,2-b:4,5-b′]dithiophene reached power conversion efficiencies (PCEs) up to 1.45% but changing the alkyl groups to more bulky ethylhexyl chains reduced the PCE to 1.17%. More noteworthy is that the photovoltaic performance improves for the polymers based on 4,4-dihexyl-4H-silolo[3,2-b:4,5-b′]dithiophene after the ester groups has been eliminated from the DTZ unit by a thermal treatment around 200 °C. To confirm that elimination of the solubilizing groups improve the long term durability of the materials the photochemical stability was estimated by a novel accelerated degradation method by which the photobleaching of the polymer was followed during degradation at 100 solar intensities. This clearly shows that the pristine polymer films are by far the most unstable under the given conditions emphasizing the unfavorable effect of solubilizing groups on the photochemical stability of conjugated polymers.
Introduction
Polymer solar cells (PSCs)1–5 distinguish themselves from all other solar cell technologies by enabling preparation under ambient conditions using low temperature roll-to-roll coating and printing6–8 of liquid polymer inks onto flexible substrates over large areas. Together with low material cost and usage these techniques allow fast deposition of polymers which can realize the presumed very low production cost of PSCs. In terms of materials for the photoactive layer in polymer solar cells low-bandgap conjugated polymers9,10 blended with a soluble fullerene derivative has proven that they can provide efficient light harvesting across a broad area of the solar spectrum. Low-bandgap polymers prepared by the donor–acceptor approach have been extensively studied over the past decade where PCEs now exceeds 8%11 for small area polymer/PCBM solar cells. In spite of promising performance, there are concerns about the photochemical stability of some of these polymer systems hence a highly efficient light harvesting polymer is of little practical consequence if its photovoltaic operation is not stable. The influence of the polymer chemical structure on its stability has been studied by Manceau et al.12 where general rules relative to the polymer structure–stability relationship are proposed. For example polymers based on benzothiadiazole and the dialkylcyclopentadithiophene unit have been widely used showing high efficiencies up to 5.5%13 but due to the presence of the quaternary bridging carbon site in the five-membered ring it can readily undergo oxidation as reported for polyfluorenes.14,15 Though recent studies have shown that substitution of the bridging carbon atom with silicon can result in a significant photochemical stability improvement of polymers by a factor 5 together with higher carrier mobility16 and enhanced interchain packing.17,18Still, a remaining issue is the fact that solubilizing groups are required in order to process polymer materials into thin films but the solubilizing groups is also a part of the reason why the polymers degrade quickly. The softness/photochemistry provided by solubilizing groups has been linked to the instability of polymer solar cells19–27 as they allow for both morphological changes along with chemical transformations caused by diffusion of small molecules and constituents. In order to overcome this setback one could explore the many possibilities in employing polymer materials bearing thermally cleavable solubilizing groups.28–33 The groups allow film formation via solution processing and can subsequently be removed in a heating step forming a harder insoluble polymer material where diffusion phenomena are slowed down. While ongoing research exploiting these novel materials has afforded promising results showing operational lifetimes well in excess of three years34 they still lack in photovoltaic performance in order to combine processability, stability and high efficiency in one material.
In this work we report our efforts in this direction through the synthesis of two novel thermally reactive copolymers based on dithienylthiazolo[5,4-d]thiazole (DTZ), bearing thermocleavable ester groups on the thienyl units, and dialkylsilolodithiophene (SDT). As reported, the use of the thiazolothiazole fused ring as electron-deficient moiety ensures a rigid coplanar backbone with extended π-electron delocalization and strong interchain stacking ability35,36 which are important attributes in highly efficient semiconductors. The bulkiness of the alkyl chains on the SDT unit proved to be very significant in terms of photovoltaic performance before and after annealing of the polymer:PCBM solar cells which is presented and discussed together with the photochemical stability.
Results and discussion
The synthetic steps involved in the preparation of the polymers are outlined in Scheme 1. The thermally reactive DTZ derivative 4 was synthesized following a known procedure37 by condensation of dithiooxamide with the aldehyde 3 giving 4 in a reasonable yield. 3-methyl-3-octyl 2-formylthiophene-3-carboxylate (3) was prepared by esterification of thiophene-3-carboxylic acid (1)28 employing 4-dimethylaminopyridine (DMAP) in combination with N,N′-diisopropylcarbodiimide (DIPC) followed by deprotonation of 2 in the 2-position using Lithium diisopropylamine (LDA) and treatment with N-formylpiperidine (NFP). Finally the DTZ derivative 4 was functionalized by bromination using N-bromosuccinimide (NBS). In order to drive the bromination to completion several equivalents NBS had to be used together with a catalytic amount of acetic acid. Copolymerisation of 5viaStille coupling with 4,4-dihexyl-2,6-bis(trimethylstannyl)-4H-silolo[3,2-b:4,5-b′]dithiophene or 4,4-bis(2-ethylhexyl)-2,6-bis(trimethylstannyl)-4H-silolo[3,2-b:4,5-b′]dithiophene gives the polymers PhxSDT-DTZ and PehSDT-DTZ in good yield (70–95%) as dark blue solids. Generally a higher molecular weight is reached for polymers carrying ethylhexyl chains on the SDT unit (Table 1) compared to regular hexyl chains which is ascribed to enhanced solubility of the bulky ethylhexyl group. At room temperature both polymers show limited solubility but dissolves in hot chlorinated solvents, PehSDT-DTZ more readily than PhxSDT-DTZ.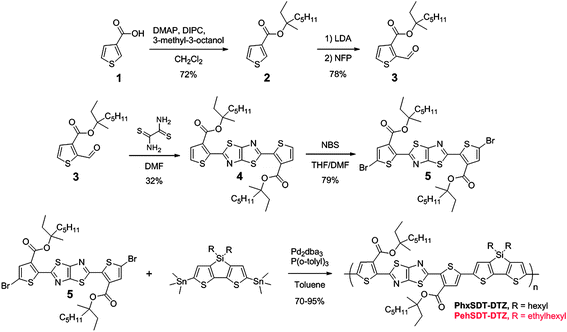 | ||
| Scheme 1 Synthetic steps involved in the preparation of the polymers | ||
| polymer | M n (g/mol) | PDI | Solution | filma | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| α (λmax) (L/g·cm) | λ max (nm) | λ onset (nm) | E g (eV) | λ max (nm) | λ onset (nm) | E g (eV) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Films annealed at 225 °C for 30 s. are indicated in parentheses. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PhxSDT-DTZ | 9500 | 3.4 | 45 | 580 | 675 | 1.84 | 580 (594) | 698 (745) | 1.78 (1.66) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PehSDT-DTZ | 14000 | 2.2 | 41 | 578 | 667 | 1.86 | 621 (603) | 690 (755) | 1.80 (1.64) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
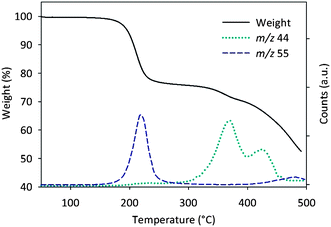
In order to investigate the thermal behavior of the polymers they were studied by thermogravimetric analysis in conjunction with mass spectrometry of the carrier gas (TGA-MS) focusing on mass signals from carbon dioxide and fragments from even and uneven alkenes. The TGA (Fig. 1) shows a ∼25% weight loss around 200 °C that together with observed fragment peaks (m/z 55) in the same temperature range corresponds well to elimination of the ester groups, as expected from earlier studies.28 Evolution of carbon dioxide (m/z 44) is only observed at temperatures well in excess of 300 °C that together with a minor weight loss can descend from decarboxylation For solar cell applications only the “low” temperature ester elimination (Scheme 2) is of interest, and thus the focus of this study, due to a higher degree of device failure and inferior reproducibility at higher temperatures.
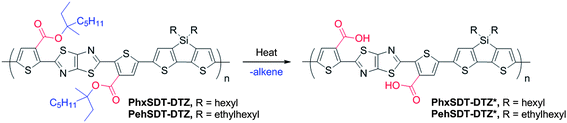 | ||
| Scheme 2 Preparation of PhxSDT-DTZ* and PehSDT-DTZ* by thermolytic elimination of ester groups | ||
The absorption spectra of the polymers in chloroform solution and in thin film have a rather similar profile (Fig. 2a) with vibronic fine structure at 600 nm, both in solution and in the solid state. The optical band gaps, defined by the onset of absorption, of the polymers in solution are ranging from 1.84–1.86 eV (Table 1). In the solid state they are red-shifted merely about 23 nm indicating a relatively high degree of interchain packing already in solution. A more significant red shift up to 88 nm can be observed when the polymer film is annealed at 225 °C (Fig. 2b). At this temperature the bulky ester groups are eliminated which most likely improve interchain association and coplanarity of the polymer thereby lowering the band gap. To check if the same trend with broadening of the absorption profile and loss of intensity after annealing (Fig. 2b) also would apply for the polymers in blends with PCBM absorption spectra of polymer/PCBM in thin film were done as well (Fig. 3). Apparently the absorption spectra of the polymer/PCBM films before and after annealing 225 °C are very similar indicating that the polymers conformation in the conjugated backbone gets locked to some extent in blends with PCBM. No clear colour change was observed subsequent to the thermal treatment though the films obtained a darker shade and was completely insoluble in organic solvents.
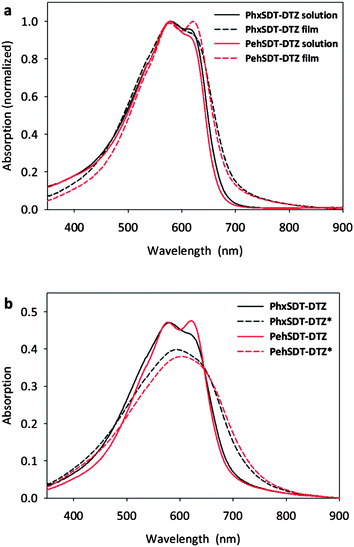 | ||
| Fig. 2 (a) UV-vis absorption spectra of the polymers in chloroform solution and (b) in thin film (pristine and annealed at 225 °C for 30 s.). Thermocleaved films are indicated with an asterisk (*). | ||
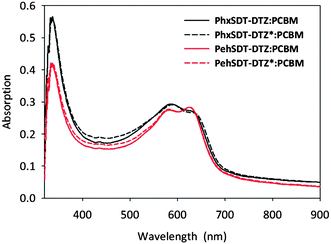 | ||
Fig. 3
UV-vis absorption spectra of polymer/PCBM (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) in thin film (pristine and annealed at 225 °C for 30 s.). Thermocleaved films are indicated with an asterisk (*). :2) in thin film (pristine and annealed at 225 °C for 30 s.). Thermocleaved films are indicated with an asterisk (*). | ||
The photovoltaic performance of the polymers were tested in bulk heterojunction solar cells with the conventional device architecture ITO/PEDOT:PSS/polymer:PCBM/Al. The optimized solar cell efficiencies are summarized in Table 2 and the obtained current–voltage curves of the polymer:PCBM solar cells are presented in Fig. 4a. High open circuit voltage (Voc) up to 0.76 V can be reached with both polymers but the best devices were prepared with PhxSDT-DTZ due to a higher current density (Jsc). Though the effect of thermocleaving is where the solar cells truly vary despite the fact that the tested polymers only differ by the nature of the alkyl chains on the SDT unit. Generally the Voc drops 0.1 V after annealing at 225 °C but together with an increase in the current density of PhxSDT-DTZ devices from 4.98 mA/cm2 up to 5.93 mA/cm2 the PCE is enhanced from 1.36% to 1.45% upon thermocleaving. For PehSDT-DTZ devices thermocleaving leads to a drop in the current density from 4.01 mA/cm2 to 3.52 mA/cm2 which reduces the PCE from 1.17% to 0.86%. For the PhxSDT-DTZ devices the IPCE (Fig. 4b) reaches 50% with a photoresponse up to 720 nm. In contrast, PehSDT-DTZ devices only reach 36% IPCE that drops to 30% upon thermocleaving whereas thermocleaved PhxSDT-DTZ* devices are enhanced 1–6% at roughly all wavelengths compared to pristine PhxSDT-DTZ devices.
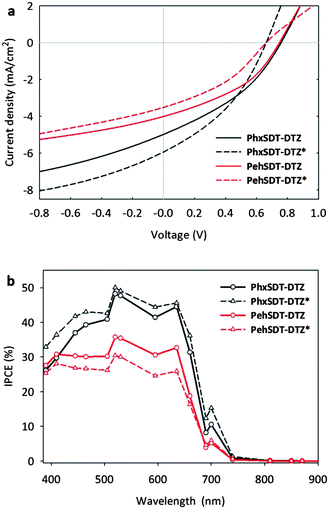 | ||
| Fig. 4 (a) J–V characteristics of polymer:PCBM solar cells measured under 100 mW/cm2 white light (b) IPCE of polymer:PCBM solar cells. | ||
The performance of bulk heterojunction polymer/PCBM solar cells is known to be very dependent on the morphology of the active layer38–40 and different factors can have an influence on the morphology including the annealing temperature.28 Changes in the surface topography of the PhxSDT-DTZ:PCBM and PehSDT-DTZ:PCBM device films were measured by atomic force microscopy (AFM) and the images are shown in Fig. 5. Earlier work have reported extensive phase segregation of thermocleavable polymers and PCBM upon thermocleaving which can possibly limit charge carrier generation and transport. Though, when comparing the device films in this study before and after thermocleaving only a minor increase of the domain sizes to larger features are observed when the films are annealed at 225 °C. The pristine polymer/PCBM films (Fig. 5. a + c) have a surface roughness ranging from 0.67–0.69 nm. After thermocleaving of the polymers (Fig. 5. b + d) the surface roughness increases 0.04 nm (PhxSDT-DTZ*:PCBM) and 0.12 nm (PehSDT-DTZ*:PCBM) respectively. The films based on PehSDT-DTZ* display the largest increase in the surface roughness by a factor 3 compared to PhxSDT-DTZ* indicating that PhxSDT-DTZ* has a more ordered polymer structure which is beneficial for the charge carrier transport. The indication that the bulky ethylhexyl groups in PehSDT-DTZ are inhibiting the ordering of the polymeric chains can partly explain the difference in the J–V characteristics of the two polymers.
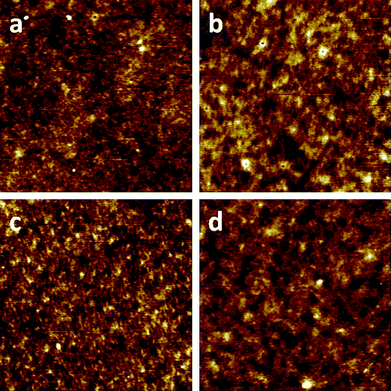 | ||
| Fig. 5 AFM topography images (5 μm × 5 μm) of solar cells based on blends of PCBM and (a) pristine PhxSDT-DTZ, Sa = 0.67 nm (b) PhxSDT-DTZ annealed at 225 °C, Sa = 0.71 nm (c) pristine PehSDT-DTZ, Sa = 0.69 nm (d) PehSDT-DTZ annealed at 225 °C, Sa = 0.81 nm. | ||
To further investigate the molecular order, Grazing Incidence Wide Angle X-ray Scattering (GIWAXS) patterns were obtained from polymer/PCBM thin films, deposited by spin coating a 1:2 blend of the polymer (PhxSDT-DTZ or PehSDT-DTZ) and PCBM dissolved in 1,2-dichlorobenzene on a silicon wafer substrate. In the wide angle scattering range, the X-ray scattering is sensitive to crystalline structure which is clearly detected for PhxSDT-DTZ (Fig. 6a) whereas PehSDT-DTZ shows very limited diffraction from the lamellar stacking, most likely forming more amorphous films due to the bulky ethylhexyl groups. The higher degree of crystallinity of PhxSDT-DTZ compared to PehSDT-DTZ corresponds well to the variation in the J–V characteristics where PhxSDT-DTZ shows the best performance of the two polymers (Table 2). There is almost no difference in the films after thermocleaving to PhxSDT-DTZ* and PehSDT-DTZ* and all films appear to have a lamellar 100 top around 0.37 Å−1 corresponding to a d-spacing of 17 Å which is very close to that of the state of the art polymer poly-3-hexylthiophene (P3HT) that packs with a lamellar spacing of 16.7° Å.41
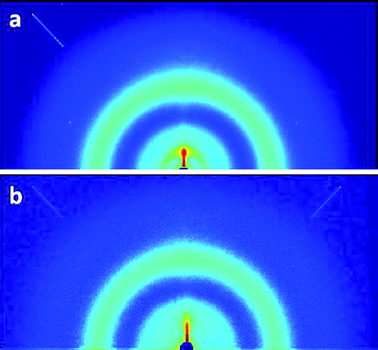 | ||
| Fig. 6 Grazing incidence wide angle X-ray scattering data as recorded for the thin films of (a) PhxSDT-DTZ/PCBM and (b) PehSDT-DTZ/PCBM with colors representing intensity on a log scale. The detector area covers up to a scattering vector modulus q ∼ 3.5 Å−1 (q = 4πsin(θ)/λ), 2θ is the scattering angle, λ is the X-ray wavelength). | ||
To rapidly evaluate the stability of the pristine polymers and the effect of thermocleaving, an accelerated stability test was performed with a novel light concentrating setup. The polymer films were spin coated on glass substrates and exposed to 100 solar intensities (10 W/cm2) while monitoring the decrease of UV-vis absorption. Fig. 7 presents the evolution of the normalized number of absorbed photons for the best performing polymer (PhxSDT-DTZ) alone and in a PCBM blend in the pristine and the annealed case. The stability of the polymers at 100 solar intensities cannot be directly converted into an expected stability at 1 sun in a conventional solar simulator. However, it provides good estimate of the relative stabilities between different polymers,42 and therefore a P3HT reference is included, since the stability of this material’s standard degradation conditions is well known. The stability of PhxSDT-DTZ is observed to be more than twice the one of the P3HT reference polymer with respect to the time it takes for the PhxSDT-DTZ film to reach half of its initial absorbance (T50), which proves the high photochemical stability of this polymer. Furthermore, the thermocleaving is found to introduce a considerable stability improvement of the polymer PhxSDT-DTZ. The T50 is 12 min which increases with 50% for the thermocleaved polymer PhxSDT-DTZ* to a T50 of 18 min. Moreover the introduction of PCBM creating a (1:2) PhxSDT-DTZ:PCBM blend approximately doubled the T50 both in the case of the pristine and the annealed films. This can be explained by the fact that PCBM can function as an effective radical scavenger43–45 thereby diminishing chemical transformations of the materials induced by formed radicals in the active layer. In addition, it has also been reported that PCBM can function as an efficient quencher of the polymer singlet state in P3HT:PCBM blends,46 which can stabilize the system further.
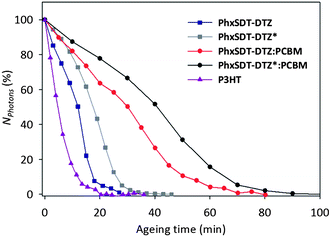 | ||
| Fig. 7 Evolution of the normalized absorption during accelerated photochemical ageing in air under 100 solar intensities. | ||
Conclusion
In conclusion, new thermally reactive copolymers based on DTZ and SDT, demonstrating band gaps in the range of 1.64–1.80 eV, have been synthesized and tested in bulk heterojunction polymer/PCBM solar cells. For improved photochemical stability, the solubilising chains on the DTZ unit are thermally removable ester groups which allow processing of two chemically different thin films from the same precursor polymer through ester elimination around 200 °C. A significant effect is observed on the photovoltaic performance after thermocleaving depending upon the bulkiness of the alkyl chains on the SDT unit. The PCE for polymers based on 4,4-dihexyl-4H-silolo[3,2-b:4,5-b']dithiophene enhanced from 1.36% to 1.45% upon thermocleaving but shifting the alkyl groups to more bulky ethylhexyl chains reduced the PCE from 1.17% to 0.86% upon thermocleaving due to a significant drop in the current density. This can partly be explained by the inhibiting effect of the ethylhexyl chains, versushexyl chains, on the ordering of the polymeric chains which might hamper the charge carrier transport. Finally, measuring the amount of absorbed photons under 100 suns versus the ageing time for PhxSDT-DTZ clearly shows that the pristine polymer films are by far the most unstable under the given conditions while the ageing time is enhanced with 50% for the thermocleaved polymer PhxSDT-DTZ* with respect to T50. Moreover, it was observed that the degradation is strongly slowed down when the polymer is blended with PCBM which has been ascribed to the effective radical scavenging properties of PCBM. The PhxSDT-DTZ*:PCBM system appeared to be very chemically stable under the given conditions and forthcoming research will focus on optimization of the photovoltaic performance of this class of material in order to realize the overall goal of processability, stability and high efficiency in one material.Experimental section
Polymer solar cell fabrication and analysis
Photovoltaic devices were made by spin coating PEDOT:PSS (Aldrich, 1.3 wt % aqueous solution) onto precleaned, patterned indium tin oxide (ITO) substrates (9–15 Ω per square) (LumTec) followed by annealing at 140 °C for 5 min. The active layer was deposited by spin coating a 1:2 blend of the polymer and PCBM dissolved in 1,2-dichlorobenzene (25 mg/ml). After spin coating of the active layer the devices were either processed directly into a solar cell by evaporation of aluminium as back electrode or subjected to a thermal treatment at 225 °C immediately before evaporation of the back electrode at 2–3 × 10−6 mbar. The active area of the cells was 0.5 cm2. I–V characteristics were measured under AM1.5G corresponding to 100 mW/cm2 white light from a multi-wavelength high-power LED array using a Keithley 2400 source meter. IPCE spectra were recorded on the same solar test platform47 with the LED based illumination system.
Photochemistry
Pure polymer samples (P3HT and PhxSDT-DTZ) and polymer:PCBM blends (PhxSDT-DTZ:PCBM) were spin-coated under air on glass slides from 1,2-dichlorobenzene solutions. The solution concentrations and spinning speeds were adjusted to get a film thickness around 100 nm. Accelerated degradation studies were performed with a novel light concentrating system. A 1200 W HMI lamp was focused onto the samples with an intensity of 10 W/cm2, which amounts to 100 solar intensities. The light was not filtered, which implies that the light was UV rich with a cutoff at 280 nm. Since keeping a constant temperature for all samples is technically complicated, all samples were allowed to reach their equilibrium temperature during the degradations. The samples were placed in a sample holder that allowed for free passage of the transmitted light, to avoid excessive heating of the samples. All degradations were made in the ambient at room temperature. Quantification of the degradation rate was based on the evolution of the gradual decrease of UV-visible absorbance, which was recorded in 30 s intervals. Tromholt et al.42 have described the degradation quantification in detail, however in the context of an alternative light concentrating setup.Acknowledgements
This work was supported by the Danish Strategic Research Council (2104-07-0022)Reference list
- P. L. Boudreault, A. Najari and M. Leclerc, Chem. Mater., 2011, 23, 456 CrossRef CAS.
- C. J. Brabec, S. Gowrisanker, J. J. M. Halls, D. Laird, S. J. Jia and S. P. Williams, Adv. Mater., 2010, 22, 3839 CrossRef CAS.
- W. Cai, X. Gong and Y. Cao, Sol. Energy Mater. Sol. Cells, 2010, 94, 114 CrossRef CAS.
- A. Facchetti, Chem. Mater., 2011, 23, 733 CrossRef CAS.
- M. Helgesen, R. Søndergaard and F. C. Krebs, J. Mater. Chem., 2010, 20, 36 RSC.
- Y. Galagan, I. G. de Vries, A. P. Langen, R. Andriessen, W. J. Verhees, S. C. Veenstra and J. M. Kroon, Chem. Eng. Process., 2011, 50, 454 CrossRef CAS.
- F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2009, 93, 394 CrossRef CAS.
- L. Wengeler, B. Schmidt-Hansberg, K. Peters, P. Scharfer and W. Schabel, Chem. Eng. Process., 2011, 50, 478 CrossRef CAS.
- E. Bundgaard and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2007, 91, 954 CrossRef CAS.
- R. Kroon, M. Lenes, J. C. Hummelen, P. W. M. Blom and B. de Boer, Polym. Rev., 2008, 48, 531 Search PubMed.
- http://www.%20solarmer.%20com/, http://www.%20konarka.%20com/, 2011.
- M. Manceau, E. Bundgaard, J. E. Carle, O. Hagemann, M. Helgesen, R. Søndergaard, M. Jørgensen and F. C. Krebs, J. Mater. Chem., 2011, 21, 4132 RSC.
- J. Peet, J. Y. Kim, N. E. Coates, W. L. Ma, D. Moses, A. J. Heeger and G. C. Bazan, Nat. Mater., 2007, 6, 497 CrossRef CAS.
- E. J. W. List, R. Guentner, P. S. de Freitas and U. Scherf, Adv. Mater., 2002, 14, 374 CrossRef CAS.
- K. L. Chan, M. J. McKiernan, C. R. Towns and A. B. Holmes, J. Am. Chem. Soc., 2005, 127, 7662 CrossRef CAS.
- J. H. Hou, H. Y. Chen, S. Q. Zhang, G. Li and Y. Yang, J. Am. Chem. Soc., 2008, 130, 16144 CrossRef CAS.
- H. Y. Chen, J. H. Hou, A. E. Hayden, H. Yang, K. N. Houk and Y. Yang, Adv. Mater., 2010, 22, 371 CrossRef CAS.
- M. Morana, H. Azimi, G. Dennler, H. J. Egelhaaf, M. Scharber, K. Forberich, J. Hauch, R. Gaudiana, D. Waller, Z. H. Zhu, K. Hingerl, S. S. van Bavel, J. Loos and C. J. Brabec, Adv. Funct. Mater., 2010, 20, 1180 CrossRef CAS.
- F. C. Krebs and K. Norrman, Progr. Photovolt.: Res. Appl., 2007, 15, 697 Search PubMed.
- M. Lira-Cantu, K. Norrman, J. W. Andreasen and F. C. Krebs, Chem. Mater., 2006, 18, 5684 CrossRef CAS.
- K. Norrman and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2006, 90, 213 CrossRef CAS.
- K. Norrman, N. B. Larsen and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2006, 90, 2793 CrossRef CAS.
- S. Chambon, A. Rivaton, J. L. Gardette and M. Firon, Sol. Energy Mater. Sol. Cells, 2008, 92, 785 CrossRef CAS.
- S. Chambon, M. Manceau, M. Firon, S. Cros, A. Rivaton and J. L. Gardette, Polymer, 2008, 49, 3288 CrossRef CAS.
- M. Manceau, A. Rivaton and J. L. Gardette, Macromol. Rapid Commun., 2008, 29, 1823 CrossRef CAS.
- M. Manceau, A. Rivaton, J. L. Gardette, S. Guillerez and N. Lemaitre, Polym. Degrad. Stab., 2009, 94, 898 CrossRef CAS.
- M. Manceau, A. Rivaton, J. L. Gardette, S. Guillerez and N. Lemaitre, Sol. Energy Mater. Sol. Cells, 2011, 95, 1315 CrossRef CAS.
- M. Helgesen, M. Bjerring, N. C. Nielsen and F. C. Krebs, Chem. Mater., 2010, 22, 5617 CrossRef CAS.
- M. Helgesen and F. C. Krebs, Macromolecules, 2010, 43, 1253 CrossRef CAS.
- M. Helgesen, S. A. Gevorgyan, F. C. Krebs and R. A. J. Janssen, Chem. Mater., 2009, 21, 4669 CrossRef CAS.
- L. H. Nguyen, S. Gunes, H. Neugebauer, N. S. Sariciftci, F. Banishoeib, A. Henckens, T. Cleij, L. Lutsen and D. Vanderzande, Sol. Energy Mater. Sol. Cells, 2006, 90, 2815 CrossRef CAS.
- H. Dilien, J. Vandenbergh, F. Banishoeb, P. Adriaensens, T. J. Cleij, L. Lutsen and D. J. M. Vanderzande, Macromolecules, 2011, 44, 711 CrossRef CAS.
- M. H. Petersen, O. Hagemann, K. T. Nielsen, M. Jørgensen and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2007, 91, 996 CrossRef CAS.
- F. C. Krebs and H. Spanggaard, Chem. Mater., 2005, 17, 5235 CrossRef CAS.
- S. Ando, J.i. Nishida, H. Tada, Y. Inoue, S. Tokito and Y. Yamashita, J. Am. Chem. Soc., 2005, 127, 5336 CrossRef CAS.
- I. Osaka, G. Sauve, R. Zhang, T. Kowalewski and R. D. Mccullough, Adv. Mater., 2007, 19, 4160 CrossRef CAS.
- D. A. Thomas, J. Heterocycl. Chem., 1970, 7, 457 CAS.
- J. K. J. van Duren, X. N. Yang, J. Loos, C. W. T. Bulle-Lieuwma, A. B. Sieval, J. C. Hummelen and R. A. J. Janssen, Adv. Funct. Mater., 2004, 14, 425 CrossRef CAS.
- X. Yang and J. Loos, Macromolecules, 2007, 40, 1353 CrossRef CAS.
- S. Barrau, V. Andersson, F. L. Zhang, S. Masich, J. Bijleveld, M. R. Andersson and O. Inganas, Macromolecules, 2009, 42, 4646 CrossRef CAS.
- T. J. Prosa, M. J. Winokur, J. Moulton, P. Smith and A. J. Heeger, Macromolecules, 1992, 25, 4364 CrossRef CAS.
- T. Tromholt, M. Manceau, M. Helgesen, J. E. Carlé and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2010, 95, 1308.
- C. N. Mcewen, R. G. Mckay and B. S. Larsen, J. Am. Chem. Soc., 1992, 114, 4412 CrossRef.
- P. J. Krusic, E. Wasserman, P. N. Keizer, J. R. Morton and K. F. Preston, Science, 1991, 254, 1183 CrossRef CAS.
- E. B. Zeynalov, N. S. Allen and N. I. Salmanova, Polym. Degrad. Stab., 2009, 94, 1183 CrossRef CAS.
- M. Manceau, S. Chambon, A. Rivaton, J. L. Gardette, S. Guillerez and N. Lemaitre, Sol. Energy Mater. Sol. Cells, 2010, 94, 1572 CrossRef CAS.
- F. C. Krebs, K. O. Sylvester-Hvid and M. Jørgensen, Prog. Photovoltaics, 2009, 19, 97.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental procedures, synthetic procedures and characterization data of the monomers and polymers according to Scheme 1. See DOI: 10.1039/c1py00323b |
| This journal is © The Royal Society of Chemistry 2011 |