KOH-catalyzed highly efficient aminohalogenation of β-nitrostyrenes with t-butyl N,N-dichlorocarbamate as nitrogen/halogen source †
Haibo
Mei
a,
Jianlin
Han
*a,
Guigen
Li
a and
Yi
Pan
*ab
aSchool of Chemistry and Chemical Engineering, Nanjing, 210093, China. E-mail: hanjl@nju.edu.cn; Fax: +86-25-83593153; Tel: +86-25-83593153
bState of Key Laboratory of Coordination, Nanjing, 210093, China. E-mail: yipan@nju.edu.cn
First published on 9th August 2011
Abstract
A highly efficient and facile aminohalogenation of β-nitrostyrenes with t-butyl N,N-dichlorocarbamate (BocNCl2) as nitrogen/halogen source is reported, which tolerated a wide scope of substrates and could be completed within 20 min at room temperature with excellent chemical yields.
Introduction
Aminohalogenation, and related reactions of functionalized olefins, has become one of the most useful and attractive tools for constructing carbon–nitrogen and carbon–halogen bonds in tandem fashion at the same time.1–4 The resulting vicinal haloamines from these reactions belong to an important class of building blocks in both organic synthesis and medicinal chemistry.5 They can also be readily converted into varieties of other intermediates by replacing the halogen atoms in both intramolecular and intermolecular substitution processes.6 In recent years, several new catalytic aminohalogenation systems have been developed for a series of functionalized alkenes, including α,β-unsaturated carboxylic esters,7α,β-unsaturated nitriles,8α,β-unsaturated ketones,9β-nitrostyrenes10 and so on.11 The nitrogen/halogen sources for these aminohalogenation processes were found to play a significant role during the formation of corresponding products. To date, several amides have been tired as nitrogen/halogen sources for aminohalogenation reaction of functionalized olefins, which usually focused on the N,N-dihalosulfonamide,12–15 and the combination of sulfonamide/NCS or NBS.16 Although some other amides have been reported to be nitrogen sources, such as succinimide/NCS,17N-bromoacetamide18 and benzamides/NCS,19 most of them suffer from the shortcoming of quite long reaction time. Furthermore, the N-protecting groups are difficult to remove from the resulted haloamine products, which greatly limits the developments and applications of such aminohalogenation reactions.β-nitrostyrenes represent a particularly important and useful class of substrates for the aminohalogenation,10,17–19 because the resulted haloamine products can be easily converted into vicinal diamines.20 In the last five years, we and other groups have reported some aminohalogenation reactions on these electron-deficient alkenes with amides as nitrogen sources.10,17–19 However, the nitrogen sources for this transformation still remain a great challenge, which usually need more than 24 h to convert the β-nitrostyrenes into the corresponding products. N,N-dichlorocarbamates have been used for aminohalogenation of simple, as well as electron deficient olefins such as α,β-unsaturated esters and nitriles.21 In particular t-butyl N,N-dichlorocarbamate has been used for aminohalogenation of simple olefins.22 In our ongoing research on aminohalogenation, we found that tert-butyl dichlorocarbamate could be used as efficient nitrogen source for aminohalogenation of β-nitrostyrenes. The advantage of such an exploration is that the aminohalogenation reaction with t-butyl N,N-dichlorocarbamate as nitrogen/chlorine source can be completed within a really short time, comparing to the previous reported N,N-dihalosulfonamide.12–15 Herein, we would like to report a facile and highly efficient aminochlorination of β-nitrostyrenes with tert-butylN,N-dichlorocarbamate (BocNCl2) as nitrogen/halogen source by using commercially available KOH as the catalyst, giving dichlorinated products with up to 99% chemical yields (Scheme 1).
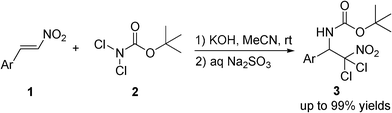 | ||
| Scheme 1 Aminohalogenation with BocNCl2. | ||
Results and discussion
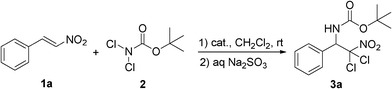
Based on the previous studies on the aminohalogenation of β-nitrostyrenes,10a the initial reaction conditions were focused on the use of β-nitrostyrene 1a with 2.5 equiv of BocNCl2 as nitrogen source in CH2Cl2 at room temperature. The reaction without any catalyst did not happen and most of the starting materials remained even after the reaction time was extended for quite a long time (entry 1, Table 1). Then, several metal catalysts (entries 2–8) and organocatalysts (entries 10–12) were tried in this reaction, but almost no desired haloamine products were observed at all. Further scan of the conditions revealed that the transformation could proceed smoothly in the presence of K2CO3, affording the desired product 3a in 72% yield after 20 h (entry 9). To our delight, KOH was found to work very well in the reaction, resulting in an excellent chemical yield (91%), as well as a really short reaction time (3 h, entry 13).|
|
|||
|---|---|---|---|
| Entry | Catalyst | Time | Yield (%)b |
| a Reaction conditions: β-nitrostyrene (0.5 mmol), BocNCl2 (1.25 mmol), catalyst (20 mol%), CH2Cl2 (5 mL). b Isolated yields. | |||
| 1 | None | 6 d | NR |
| 2 | Mn(OAc)2 | 6 d | <5 |
| 3 | Ni(OAc)2 | 6 d | <10 |
| 4 | CuCl | 6 d | <5 |
| 5 | CuI | 6 d | <5 |
| 6 | Pd(OAc)2 | 6 d | NR |
| 7 | NiCl2 | 6 d | <10 |
| 8 | PdCl2 | 6 d | NR |
| 9 | K2CO3 | 20 h | 72 |
| 10 | DMAP | 6 d | <5 |
| 11 | Ph3P | 6 d | NR |
| 12 | 2,2′-Bpy | 6 d | NR |
| 13 | KOH | 3 h | 91 |
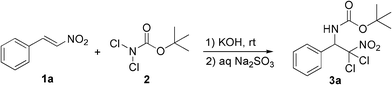
Encouraged by the excellent result with KOH as catalyst, the scan of various organic solvents was carried out to improve the reaction efficiency (Table 2). The solvent was found to be crucial for this transformation, and showed great effects on the chemical yields, as well as the reaction time. The reaction with THF as solvent could be completed in 40 min, but gave a lower yield (78%, entry 2). Acetonitrile was found to be the best choice. The reaction proceeded very quickly and was completed in quite a short time (15 min), resulting in a higher chemical yield (93%, entry 5). To the best of our knowledge, this is the shortest time for the aminohalogenation of functionalized olefins until now.7–12,16 As shown in Table 2, we found that THF and acetonitrile could clearly accelerate the reaction rate, which disclosed that they play an important role in the reaction. It was assumed that THF could stabilize the chloronium intermediates, while acetonitrile could activate the potassium hydroxide catalyst.
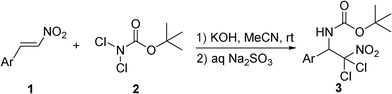
After obtaining the optimized reaction conditions, varieties of β-nitrostyrenes were used as substrates to investigate the scope of the current system (Table 3). As shown in Table 3, almost all of the tested substrates worked well in this reaction. Generally, β-nitrostyrenes with substituents on the meta-position or para-position of the aromatic ring could participate better in the reaction and usually completed the reaction within 20 min. The substituent groups on the aromatic rings did not show great effects on the reaction efficiency. Both electron-rich (entries 8 and 9) and electron-deficient (entries 5–7 and 10–13) substrates were found to be suitable for the current reaction, giving excellent chemical yields (90%–99%), even though the substituent groups were fluoro (entries 5 and 10) or trifluoromethyl (entry 13). However, the substrates with the ortho-substituted aromatic ring gave lower yields (entries 2–4) and usually needed longer reaction time to consume all the starting materials (8 h, entries 2 and 3). Notably, β-nitrostyrene with double substituted aromatic ring 1o was also well tolerated in this reaction along with a good chemical yield (79%, entry 15). It was also found that 1-naphthyl β-nitrostyrene 1p worked well in the current system, and only 15 min were needed to complete the reaction with a moderate yield (entry 16).
|
|
||||
|---|---|---|---|---|
| Entry | Ar | Time | Product | Yield (%)b |
| a Reaction conditions: β-nitrostyrene (0.5 mmol), BocNCl2 (1.25 mmol), KOH (20 mol%), MeCN (5 mL). b Isolated yields. | ||||
| 1 | C6H5 | 15 min | 3a | 93 |
| 2 | 2-CH3C6H4 | 8 h | 3b | 77 |
| 3 | 2-ClC6H4 | 8 h | 3c | 75 |
| 4 | 2-OBnC6H4 | 20 min | 3d | 76 |
| 5 | 3-FC6H4 | 20 min | 3e | 93 |
| 6 | 3-ClC6H4 | 10 min | 3f | 92 |
| 7 | 3-BrC6H4 | 15 min | 3g | 90 |
| 8 | 4-CH3C6H4 | 15 min | 3h | 99 |
| 9 | 4-OCH3C6H4 | 20 min | 3i | 96 |
| 10 | 4-FC6H4 | 12 min | 3j | 93 |
| 11 | 4-ClC6H4 | 13 min | 3k | 95 |
| 12 | 4-BrC6H4 | 14 min | 3l | 94 |
| 13 | 4-CF3C6H4 | 15 min | 3m | 91 |
| 14 | 4-CNC6H4 | 20 min | 3n | 74 |
| 15 | 3-Br,4-OCH3C6H3 | 17 min | 3o | 79 |
| 16 | 1-naphthyl | 15 min | 3p | 56 |
The trends of the reaction rate have also been found in the current aminohalogenation reaction of β-nitrostyrene derivatives. As shown in Table 3, the reaction time for the substrates with ortho-substituted aromatic ring was longer than those of the substituents on other positions (entries 2–4). It was mainly because the steric hindrance of the substrates slowed down the addition of the nitrogen/chlorine source.
The structure of these dichlorinated haloamides has been confirmed by the X-ray diffraction analysis of 3a (Fig. 1).
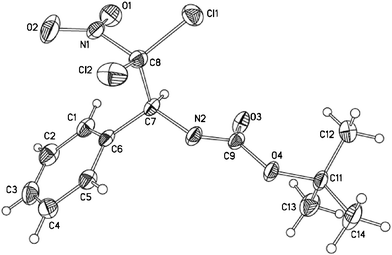 | ||
| Fig. 1 ORTEP diagram showing compound 3a. | ||
Then, we tried to raise a more simple system for such aminohalogenation of β-nitrostyrenes. The combination of BocNH2/NCS was employed as nitrogen/halogen source (Scheme 2), instead of BocNCl2, for the current system, which was similar to the previous reported TsNH2/NBS system for the aminohalogenation reaction.16 Although the combination of BocNH2/NCS also can work as nitrogen/halogen source in the current system and form the desired haloamine product, the yield was lower, and 12 h were needed for the consumption of the starting materials.
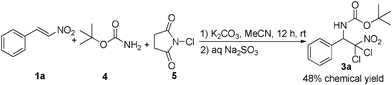 | ||
| Scheme 2 Aminohalogenation with BocNH2/NCS. | ||
According to a previous report10a on the aminohalogenation of β-nitrostyrene and the reactivity of haloamide and halocarbamate towards nucleophiles,23 a proposed pathway involving the predominant formation of chloronium intermediates is offered in Scheme 3 for this aminohalogenation process. In the initial step, BocNCl2 is activated by KOH, forming the intermediates A1 and A2. Then, A1 adds to β-nitrostyrene 1a, generating the chloronium intermediate B1, along with the transformation from A2 to B2. The chloronium intermediate B1 undergoes a ring-opening process, attacked by BocNCl−B2 on its α-position which is more positively charged than its β-position, resulting in intermediate C, a normal monohaloamino product. The final step is the formation of dichlorinated compound Dvia deprotonation/electrophilic chlorination of precursor C. The final product 3a is obtained by treatment with aqueous Na2SO3.
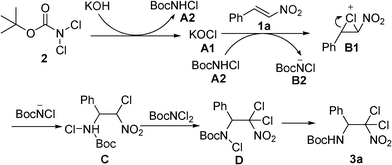 | ||
| Scheme 3 Proposed mechanism for the current process. | ||
Experimental section
General information
All aminohalogenation reactions were performed in oven-dried vials under a N2 atmosphere. The solvent acetonitrile was dried and distilled prior to use. BocNCl223 was prepared according to reported methods. The other chemicals were used as obtained from commercial sources without further purification. Flash chromatography was performed using silica gel 60 (200–300 mesh). Thin layer chromatography was carried out on silica gel 60 F-254 TLC plates of 20 cm × 20 cm. Melting points are uncorrected. IR spectra were collected on Bruker Vector 22 in KBr pellets. 1H and 13C NMR (TMS used as internal standard) spectra were recorded with a Bruker ARX 300 spectrometer. High resolution mass spectra for all the new compounds were done by a Micromass Q-Tof instrument (ESI).Aminohalogenation of β-nitrostyrenes with BocNCl2
Into an oven-dried reaction vial flushed with N2 were added β-nitrostyrenes (0.5 mmol), KOH (0.1 mmol), anhydrous MeCN (5.0 mL) and BocNCl2 (1.25 mmol). The reaction mixture was stirred at room temperature for a desired time, and then the reaction was quenched with saturated Na2SO3 (3.0 mL). The organic layer was taken and the aqueous layer was extracted with EtOAc (2 × 20 mL). The combined organic layers were dried with anhydrous Na2SO4, filtered and the solvent was removed to give the crude product, which was purified by TLC plate (hexane/EtOAc, 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).
:1).
Tert-butyl 2,2-dichloro-2-nitro-1-phenylethylcarbamate (3a)
White solid, yield 93%, mp 105–107 °C. 1H NMR (CDCl3, 300 MHz): δ = 7.42 (s, 5 H), 5.99 (d, J = 9.6 Hz, 1 H), 5.57 (d, J = 9.9 Hz, 1 H), 1.45 (s, 9 H). 13C NMR (CDCl3, 75 MHz): δ = 154.0, 133.1, 129.7, 128.9, 128.7, 116.0, 81.4, 63.8, 28.2. IR (KBr): ν = 3415, 2971, 1705, 1572, 1510, 1241, 709 cm−1. HRMS [M+Na+]: calcd for C13H16N2O4Cl2Na: 357.0379, found: 357.0387.Tert-butyl 2,2-dichloro-2-nitro-1-o-tolylethylcarbamate (3b)
White solid, yield 77%, mp 154–156 °C. 1H NMR (CDCl3, 300 MHz): δ = 7.49 (d, J = 6.9 Hz, 1 H), 7.26–7.36 (m, 3 H), 6.39 (d, J = 9.9 Hz, 1 H), 5.49 (d, J = 9.9 Hz, 1 H), 2.61 (s, 3 H), 1.43 (s, 9 H). 13C NMR (CDCl3, 75 MHz): δ = 154.0, 138.1, 133.0, 131.1, 129.4, 126.5, 125.4, 116.4, 81.4, 58.6, 28.1, 20.3. IR (KBr): ν = 3260, 2987, 1709, 1585, 1367, 1155, 1019, 740 cm−1. HRMS [M+Na+]: calcd for C14H18N2O4Cl2Na: 371.0536, found: 371.0531.Tert-butyl 2,2-dichloro-1-(2-chlorophenyl)-2-nitroethylcarbamate (3c)
White solid, yield 75%, mp 167–169 °C. 1H NMR (CDCl3, 300 MHz): δ = 7.48–7.51 (m, 2 H), 7.33–7.40 (m, 2 H), 6.75 (d, J = 10.2 Hz, 1 H), 5.56 (s, 1 H), 1.44 (s, 9 H). 13C NMR (CDCl3, 75 MHz): δ = 153.6, 135.6, 132.3, 130.7, 130.2, 128.6, 127.3, 115.6, 81.6, 59.2, 28.1. IR (KBr): ν = 3258, 3148, 2992, 1708, 1585, 1367, 1155, 749 cm−1. HRMS [M+Na+]: calcd for C13H15N2O4Cl3Na: 390.9990, found: 390.9992.Tert-butyl 1-(2-(benzyloxy)phenyl)-2,2-dichloro-2-nitroethylcarbamate (3d)
White solid, yield 76%, mp 113–114 °C. 1H NMR (CDCl3, 300 MHz): δ = 7.35–7.51 (m, 7 H), 7.04 (t, J = 4.2 Hz, 2 H), 6.36 (d, J = 10.2 Hz, 1 H), 6.19 (d, J = 10.5 Hz, 1 H), 5.17 (s, 2 H), 1.44 (s, 9 H). 13C NMR (CDCl3, 75 MHz): δ = 156.9, 154.3, 136.2, 131.9, 130.9, 129.8, 128.8, 128.2, 127.4, 121.1, 116.6, 112.8, 80.9, 70.6, 61.1, 28.2. IR (KBr): ν = 3255, 2979, 1711, 1584, 1368, 1254, 749 cm−1. HRMS [M+Na+]: calcd for C20H22N2O5Cl2Na: 463.0798, found: 463.0801.Tert-butyl 2,2-dichloro-1-(3-fluorophenyl)-2-nitroethylcarbamate (3e)
White solid, yield 93%, mp 116–117 °C. 1H NMR (CDCl3, 300 MHz): δ = 7.36–7.43 (m, 1 H), 7.10–7.24 (m, 3 H), 5.99 (d, J = 9.9 Hz, 1 H), 5.55 (s, 1 H), 1.45 (s, 9 H). 13C NMR (CDCl3, 75 MHz): δ = 164.2, 160.9, 153.9, 135.5 (d, J =6.3 Hz), 130.3 (d, J =7.9 Hz), 124.8 (d, J =2.8 Hz), 116.9 (d, J =20.7 Hz), 116.1 (d, J =22.7 Hz), 81.7, 63.3, 28.1. IR (KBr): ν = 3255, 3146, 2987, 1698, 1585, 1368, 1158, 724 cm−1. HRMS [M+Na+]: calcd for C13H15N2O4Cl2FNa: 375.0285, found: 375.0292.Tert-butyl 2,2-dichloro-1-(3-chlorophenyl)-2-nitroethylcarbamate (3f)
White solid, yield 92%, mp 136–137 °C. 1H NMR (CDCl3, 300 MHz): δ = 7.38–7.44 (m, 2 H), 7.35 (d, J = 6.3 Hz, 2 H), 5.98 (d, J = 9.9 Hz, 1 H), 5.57 (s, 1H), 1.45 (s, 9 H). 13C NMR (CDCl3, 75 MHz): δ = 153.9, 135.1, 134.6, 130.9, 129.9, 129.0, 127.3, 115.4, 81.7, 63.2, 28.1. IR (KBr): ν = 3252, 3149, 2985, 1703, 1588, 1370, 1158, 722 cm−1. HRMS [M+Na+]: calcd for C13H15N2O4Cl3Na: 390.9990, found: 390.9989.Tert-butyl 1-(3-bromophenyl)-2,2-dichloro-2-nitroethylcarbamate (3g)
White solid, yield 90%, mp 153–154 °C. 1H NMR (CDCl3, 300 MHz): δ = 7.54–7.60 (m, 2 H), 7.39 (d, J = 7.5 Hz, 1 H), 7.31 (d, J = 7.8 Hz, 1 H), 5.97 (d, J = 9.9 Hz, 1 H), 5.59 (d, J = 9.6 Hz, 1 H), 1.45 (s, 9 H). 13C NMR (CDCl3, 75 MHz): δ = 153.8, 135.4, 132.8, 131.8, 130.2, 127.7, 122.7, 115.4, 81.8, 63.2, 28.1. IR (KBr): ν = 3250, 3149, 2983, 1702, 1586, 1369, 1158 cm−1. HRMS [M+Na+]: calcd for C13H15N2O4Cl2BrNa: 436.9461, found: 436.9470.Tert-butyl 2,2-dichloro-2-nitro-1-p-tolylethylcarbamate (3h)
White solid, yield 99%, mp 108–109 °C. 1H NMR (CDCl3, 300 MHz): δ = 7.32 (d, J = 8.1 Hz, 2 H), 7.23 (d, J = 8.1 Hz, 2 H), 5.94 (d, J = 9.9 Hz, 1 H), 5.54 (d, J = 10.2 Hz, 1 H), 2.38 (s, 3 H), 1.45 (s, 9 H). 13C NMR (CDCl3, 75 MHz): δ = 154.1, 139.7, 130.1, 129.4, 128.7, 116.2, 81.4, 63.6, 28.2, 21.2. IR (KBr): ν = 3260, 3153, 2981, 1707, 1586, 1367, 1155, 775 cm−1. HRMS [M+Na+]: calcd for C14H18N2O4Cl2Na: 371.0536, found: 371.0525.Tert-butyl 2,2-dichloro-1-(4-methoxyphenyl)-2-nitroethylcarbamate (3i)
White solid, yield 96%, mp 114–115 °C. 1H NMR (CDCl3, 300 MHz): δ = 7.35 (d, J = 8.7 Hz, 2 H), 6.93 (d, J = 8.7 Hz, 2 H), 5.93 (d, J = 9.6 Hz, 1 H), 5.51 (d, J = 9.9 Hz, 1 H), 3.83 (s, 3 H), 1.45 (s, 9 H). 13C NMR (CDCl3, 75 MHz): δ = 160.5, 154.0, 130.1, 125.0, 116.2, 114.0, 81.4, 63.4, 55.3, 28.2. IR (KBr): ν = 3310, 2976, 1694, 1510, 1243, 1170, 841 cm−1. HRMS [M+Na+]: calcd for C14H18N2O5Cl2Na: 387.0485, found: 387.0474.Tert-butyl 2,2-dichloro-1-(4-fluorophenyl)-2-nitroethylcarbamate (3j)
White solid, yield 93%, mp 109–110 °C. 1H NMR (CDCl3, 300 MHz): δ = 7.40–7.45 (m, 2 H), 7.06–7.14 (m, 2 H), 5.98 (d, J = 9.6 Hz, 1 H), 5.56 (d, J = 8.4 Hz, 1 H), 1.44 (s, 9 H). 13C NMR (CDCl3, 75 MHz): δ = 165.0 (d, J = 248.0 Hz), 153.9, 131.8, 130.8 (d, J = 8.5 Hz), 129.1, 115.9 (d, J = 21.4 Hz), 81.6, 63.1, 28.1. IR (KBr): ν = 3289, 2977, 1687, 1587, 1510, 1164, 839 cm−1. HRMS [M+Na+]: calcd for C13H15N2O4Cl2FNa: 375.0285, found: 375.0290.Tert-butyl 2,2-dichloro-1-(4-chlorophenyl)-2-nitroethylcarbamate (3k)
White solid, yield 95%, mp 101–103 °C. 1H NMR (CDCl3, 300 MHz): δ = 7.38 (s, 4 H), 5.97 (d, J = 9.9 Hz, 1 H), 5.56 (s, 1 H), 1.44 (s, 9 H). 13C NMR (CDCl3, 75 MHz): δ = 153.9, 135.9, 131.7, 130.2, 128.9, 115.5, 81.7, 63.2, 28.1. IR (KBr): ν = 3258, 3153, 2982, 1705, 1586, 1368, 1155, 751 cm−1. HRMS [M+Na+]: calcd for C13H15N2O4Cl3Na: 390.9990, found: 390.9998.Tert-butyl 1-(4-bromophenyl)-2,2-dichloro-2-nitroethylcarbamate (3l)
White solid, yield 94%, mp 106–107 °C. 1H NMR (CDCl3, 300 MHz): δ = 7.56 (d, J = 8.4 Hz, 2 H), 7.33 (d, J = 8.4 Hz, 2 H), 5.96 (d, J = 9.6 Hz, 1 H), 5.56 (d, J = 8.1 Hz, 1 H), 1.44 (s, 9 H). 13C NMR (CDCl3, 75 MHz): δ = 153.8, 132.2, 131.9, 130.5, 124.1, 115.4, 81.7, 63.2, 28.1. IR (KBr): ν = 3263, 3159, 2987, 1703, 1588, 1368, 1158, 1013 cm−1. HRMS [M+Na+]: calcd for C13H15N2O4Cl2BrNa: 436.9461, found: 436.9465.Tert-butyl 2,2-dichloro-2-nitro-1-(4-(trifluoromethyl)phenyl)ethylcarbamate (3m)
White solid, yield 91%, mp 117–118 °C. 1H NMR (CDCl3, 300 MHz): δ = 7.70 (d, J = 7.5 Hz, 2 H), 7.59 (d, J = 7.8 Hz, 2 H), 6.05 (d, J = 6.9 Hz, 1 H), 5.57 (d, J = 8.1 Hz, 1 H), 1.45 (s, 9 H). 13C NMR (CDCl3, 75 MHz): δ = 153.9, 137.1, 132.1 (d, J = 32.9 Hz), 129.4, 125.7 (d, J = 3.2 Hz), 121.8, 115.1, 81.9, 63.3, 28.1. IR (KBr): ν = 3327, 2982, 1686, 1589, 1327, 1166, 1071 cm−1. HRMS [M+Na+]: calcd for C14H15N2O4Cl2F3Na: 425.0253, found: 425.0249.Tert-butyl 2,2-dichloro-1-(4-cyanophenyl)-2-nitroethylcarbamate (3n)
White solid, yield 74%, mp 129–130 °C. 1H NMR (CDCl3, 300 MHz): δ = 7.74 (d, J = 8.4 Hz, 2 H), 7.60 (d, J = 8.1 Hz, 2 H), 6.05 (d, J = 10.2 Hz, 1 H), 5.58 (d, J = 9.3 Hz, 1 H), 1.44 (s, 9 H). 13C NMR (CDCl3, 75 MHz): δ = 153.8, 138.3, 132.3, 129.9, 118.0, 114.9, 113.7, 82.0, 63.3, 28.1. IR (KBr): ν = 3368, 3328, 2974, 2235, 1707, 1592, 1504, 1246, 1167, 838 cm−1. HRMS [M+Na+]: calcd for C14H15N3O4Cl2Na: 382.0332, found: 382.0330.Tert-butyl 1-(3-bromo-4-methoxyphenyl)-2,2-dichloro-2-nitroethylcarbamate (3o)
White solid, yield 79%, mp 169–171 °C. 1H NMR (CDCl3, 300 MHz): δ = 7.62 (d, J = 2.4 Hz, 1 H), 7.35 (dd, J = 2.4, 8.4 Hz, 1 H), 6.92 (d, J = 8.4 Hz, 1 H), 5.91 (d, J = 9.3 Hz, 1 H), 5.50 (d, J = 9.3 Hz, 1 H), 3.93 (s, 3 H), 1.45 (s, 9 H). 13C NMR (CDCl3, 75 MHz): δ = 156.9, 153.9, 133.4, 129.5, 126.5, 115.7, 111.9, 111.6, 81.7, 62.8, 56.3, 28.1. IR (KBr): ν = 3271, 2984, 1698, 1585, 1367, 1163, 1020, 901, 775 cm−1. HRMS [M+Na+]: calcd for C14H17N2O5Cl2BrNa: 466.9567, found: 466.9562.Tert-butyl 2,2-dichloro-1-(naphthalen-1-yl)-2-nitroethylcarbamate (3p)
White solid, yield 56%, mp 197–199 °C. 1H NMR (CDCl3, 300 MHz): δ = 8.41 (d, J = 7.8 Hz, 1 H), 7.91–7.97 (m, 2 H), 7.64–7.73 (m, 2 H), 7.52–7.60 (m, 2 H), 7.06 (d, J = 9.9 Hz, 1 H), 5.69 (d, J = 9.0 Hz, 1 H), 1.42 (s, 9 H). 13C NMR (CDCl3, 75 MHz): δ = 154.1, 133.7, 132.0, 130.7, 130.4, 129.0, 127.3, 126.3, 125.3, 125.0, 123.3, 116.4, 81.5, 57.5, 28.1. IR (KBr): ν = 3242, 3137, 2974, 1694, 1585, 1364, 1156, 776 cm−1. HRMS [M+Na+]: calcd for C17H18N2O4Cl2Na: 407.0536, found: 407.0525.Aminohalogenation of β-nitrostyrene with BocNH2 and NCS
Into an oven-dried reaction vial flushed with N2 were added β-nitrostyrenes (0.5 mmol), K2CO3 (0.1 mmol), BocNH2 (1.5 mmol), NCS (1.5 mmol) and anhydrous MeCN (5.0 mL). The reaction mixture was stirred at room temperature for 12 h, and then the reaction was quenched with saturated Na2SO3 (3.0 mL). The organic layer was taken and the aqueous layer was extracted with EtOAc (2 × 20 mL). The combined organic layers were dried with anhydrous Na2SO4, filtered and the solvent was removed to give the crude product, which was purified by TLC plate (hexane/EtOAc, 8:1).
Conclusions
In conclusion, a new system for the aminohalogenation of β-nitrostyrenes has been developed by using BocNCl2 as a new nitrogen/halogen source catalyzed by KOH in acetonitrile at room temperature. This reaction proceeded very fast and could finish within 20 min, tolerating a wide scope of substrates with good to excellent yields. Further study on aminohalogenation of this nitrogen source is presently under progress.Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (No. 20772056) and the Fundamental Research Funds for the Central Universities (No. 1107020522 and No. 1082020502). The Jiangsu 333 program (for Pan) was also acknowledged.References
- J. E. Kemp, In Comprehensive Organic Synthesis, Vol. 3; B. M. Trost and I. Fleming, ed.; Pergamon Press: Oxford, 1991, 469–513 Search PubMed.
- (a) Y. Y. Yeung, X. Gao and E. J. Corey, J. Am. Chem. Soc., 2006, 128, 9644 CrossRef CAS; (b) D. A. Griffith and S. J. Danishefsky, J. Am. Chem. Soc., 1991, 113, 5863 CrossRef CAS; (c) J. Lessard, H. Driguez and J. P. Vermes, Tetrahedron Lett., 1970, 11, 4887 CrossRef.
- (a) F. A. Daniher and P. E. Butler, J. Org. Chem., 1968, 33, 4336 CrossRef CAS; (b) F. A. Daniher and P. E. Butler, J. Org. Chem., 1968, 33, 2637 CrossRef CAS; (c) F. A. Daniher, M. T. Melchior and P. E. Butler, Chem.Commun., 1968, 931 Search PubMed.
- For recent review on aminohalogenation, see: G. Li, S. R. S. Saibabu Kotti and C. Timmons, Eur. J. Org. Chem., 2007, 2745 Search PubMed.
- J. Qui and R. B. Silverman, J. Med. Chem., 2000, 43, 706 CrossRef CAS.
- (a) D. Chen, C. Timmons, L. Guo, X. Xu and G. Li, Synthesis, 2004, 2749; (b) D. Chen, L. Guo, J. Liu, S. Kirtane, J. F. Cannon and G. Li, Org. Lett., 2005, 7, 921 CrossRef CAS; (c) H. B. Mei, L. J. Yan, J. L. Han, G. Li and Y. Pan, Chem. Biol. Drug Des., 2010, 76, 392 CrossRef CAS.
- (a) G. Li, H. X. Wei, S. H. Kim and M. Neighbors, Org. Lett., 1999, 1, 395 CrossRef CAS; (b) H. X. Wei, S. H. Kim and G. Li, Tetrahedron, 2001, 57, 3869 CrossRef CAS; (c) G. Li, H. X. Wei and S. H. Kim, Tetrahedron, 2001, 57, 8407 CrossRef.
- J. L. Han, S. J. Zhi, L. Y. Wang, Y. Pan and G. Li, Eur. J. Org. Chem., 2007, 1332 CrossRef CAS.
- (a) H. Sun, G. Q. Zhang, S. J. Zhi, J. L. Han, G. Li and Y. Pan, Org. Biomol. Chem., 2010, 8, 4236 RSC; (b) D. Chen, C. Timmons, S. Chao and G. Li, Eur. J. Org. Chem., 2004, 3097 CrossRef CAS; (c) Z. G. Chen, J. F. Wei, R. T. Li, X. Y. Shi and P. F. Zhao, J. Org. Chem., 2009, 74, 1371 CrossRef CAS.
- (a) S. J. Zhi, J. L. Han, C. Lin, G. H. An, Y. Pan and G. Li, Synthesis, 2008, 1570 CAS.
- Q. Li, M. Shi, C. Timmons and G. Li, Org. Lett., 2006, 8, 625 CrossRef CAS.
- (a) J. Liu, Y. Wang and G. Li, Eur. J. Org. Chem., 2006, 3112 CrossRef CAS; (b) I. B. Rozentsveig, G. G. Levkovskaya, T. N. Rybalova and A. N. Mirskova, Russ. J. Org. Chem., 2001, 37, 87 CrossRef CAS.
- G. G. Levkovskaya, E. V. Rudyakova, I. B. Rozentsveig, A. N. Mirskova and A. I. Albanov, Russ. J. Org. Chem., 2000, 36, 1338 CAS.
- (a) A. Yamasaki, H. Terauchi and S. Takemura, Chem. Pharm. Bull., 1976, 24, 2841 CAS; (b) H. Terauchi, A. Yamasaki and S. Takemura, Chem. Pharm. Bull., 1975, 23, 3162 CAS; (c) L. Revesz, E. Blum and R. Wicki, Tetrahedron Lett., 2005, 46, 5577 CrossRef CAS; (d) R. Shen and X. Huang, J. Org. Chem., 2007, 72, 3961 CrossRef CAS.
- (a) S. Minakata, Y. Yoneda, Y. Oderaotoshi and M. Komatsu, Org. Lett., 2006, 8, 967 CrossRef CAS; (b) G. W. Wang and X. L. Wu, Adv. Synth. Catal., 2007, 349, 1977 CrossRef CAS.
- (a) For the selected examples on the aminohalogenation with TsNH2/NBS as nitrogen source, see: V. V. Thakur, S. K. Talluri and A. Sudalai, Org. Lett., 2003, 5, 861 Search PubMed; (b) X. L. Wu, J. J. Xia and G. W. Wang, Org. Biomol. Chem., 2008, 6, 548 RSC; (c) J. F. Wei, L. H. Zhang, Z. G. Chen, X. Y. Shi and J. J. Cao, Org. Biomol. Chem., 2009, 7, 3280 RSC; (d) Z. Wang, Y. M. Zhang, H. Fu, Y. Y. Jiang and Y. F. Zhao, Synlett, 2008, 2667 CAS.
- S. J. Zhi, H. Sun, C. Lin, G. Q. Zhang, G. Li and Y. Pan, Sci. China Chem., 2010, 53, 140 Search PubMed.
- Z. G. Chen, Y. Wang, J. F. Wei, P. F. Zhao and X. Y. Shi, J. Org. Chem., 2010, 75, 2085 CrossRef CAS.
- S. J. Zhi, H. B. Mei, G. Q. Zhang, H. Sun, J. L. Han, G. Li and Y. Pan, Sci. China Chem., 2010, 53, 1946 Search PubMed.
- (a) D. Enders and J. Wiedemann, Synthesis, 1996, 1443 CrossRef CAS; (b) D. Lucet, L. Toupet, T. L. Gall and G. Mioskowski, J. Org. Chem., 1997, 62, 2682 CrossRef CAS.
- (a) T. A. Foglia and D. Swern, J. Org. Chem., 1966, 31, 3625 CrossRef CAS; (b) T. A. Foglia and D. Swern, J. Org. Chem., 1968, 33, 766 CrossRef CAS; (c) B. Crookes, T. P. Seden and R. W. Turner, Chem.Commun., 1968, 324 Search PubMed.
- B. S. Orlek and G. Stemp, Tetrahedron Lett., 1991, 32, 4045 CrossRef CAS.
- R. E. White and P. Kovacic, J. Am. Chem. Soc., 1975, 97, 1180 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, full spectroscopic data for new compounds, single-crystal X-ray diffraction analysis of 3a and copies of 1H NMR and 13C NMR spectra. CCDC reference number 820793. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1ra00174d |
| This journal is © The Royal Society of Chemistry 2011 |