Control of synthesis and optical properties of DNA templated silver nanoclusters by varying DNA length and sequence†
Guo-Yu
Lan
,
Wei-Yu
Chen
and
Huan-Tsung
Chang
*
Department of Chemistry, National Taiwan University, 1, Section 4, Roosevelt Road, Taipei, 10617, Taiwan. E-mail: changht@ntu.edu.tw; Fax: 011-886-2-3366-1171; Tel: 011-886-2-3366-1171
First published on 30th August 2011
Abstract
We have used a simple method to prepare five fluorescent Ag nanoclusters (NCs) through the NaBH4-mediated reduction of Ag+ ions in the presence of various DNA scaffolds. The emission intensities and wavelengths (536–644 nm) of the as-prepared DNA–Ag NCs were dependent on the sequence and length of the DNA scaffold. Electrospray ionization mass spectrometry of the DNA–Ag NCs revealed that different numbers of Ag atoms (2–6 atoms) were present per DNA scaffold, depending on the number and position of the cytosine bases. Using the oligonucleotide 5′-CCC(TTCC)2TT(CCAA)2CCC-3′ (DNATAr2) as the scaffold, we obtained DNATAr2–Ag NCs exhibiting a quantum yield (Φf) of 61% at 608 nm; these NCs were stable in the presence of the tested thiols, Cl− ions and DNase I. Because of their strong fluorescence and stability, the DNATAr2–Ag NCs were highly selective and sensitive for the detection of Hg2+ ions [linear range: 2.5–50 nM; limit of detection (signal-to-noise ratio = 3): 0.9 nM]. We validated the practicality of this probe through analyses of several water samples spiked with Hg2+ ions (10 nM); the recoveries were 98–118%.
Introduction
Metal nanomaterials comprising up to tens of gold (Au) or silver (Ag) atoms are interesting materials because of their unusual optical and catalytic properties.1,2 For example, Au and Ag nanodots (NDs) and nanoclusters (NCs) have high fluorescence quantum yields (Φf) because their low density of states favors radiative relaxation from electronically excited states.3 Along with their long Stokes shifts, bio-compatibility, and stability, such Au and Ag NDs/NCs have attracted considerable attention for their use in the sensing of analytes and for cell imaging.4Relative to Au NCs and Au NDs,4a,bAg NCs usually exhibit greater values of Φf, but they are slightly less stable. Ag NCs can be prepared through sodium borohydride (NaBH4)-mediated reduction of Ag+ ions in the presence of polymeric templates [e.g., poly(N-isopropylacrylamide-acrylic acid-2-hydroxyethyl acrylate), poly(amidoamine), and DNA].1,5,6 Because DNA templates are highly hydrophilic and have strong affinity for Ag+ ions, they have become popular for the preparation of DNA–Ag NCs ever since the pioneering studies of Dickson and coworkers.1a Mass spectrometric analysis has revealed that DNA–Ag NCs typically feature 2–10 Ag atoms in each DNA scaffold.7 Furthermore, 1H NMR spectroscopy has revealed that Ag ions (atoms) have high affinity toward cytosine (C) bases through C–Ag–C coordination.1a Varying the length and sequence of the DNA template allows ready tuning of the optical properties of the DNA–Ag NCs.1,7a,8 For example, the emission wavelength of DNA–Ag NCs can be tuned from blue to the near-infrared merely by varying the sequence of a 12-base DNA template.1c These water-soluble DNA–Ag NCs exhibit excellent photostability, high values of Φf, and essentially no photoblinking within a period of 1 s.9 Nevertheless, their stability in high-salt media (e.g., physiological conditions) remains poor.7a In addition, the detection of trace amounts of analytes requires highly fluorescent DNA–Ag NCs.
In this study, we prepared five different DNA–Ag NCs that emitted at wavelengths ranging from 536 to 644 nm. We investigated the effects of the length and sequence of the DNA templates on the sizes and optical properties of the DNA–Ag NCs, and monitored the thiol-induced fluorescence quenching and stability of the as-prepared DNA–Ag NCs against nuclease degradation. Finally, we employed the DNA–Ag NCs for the sensitive and selective detection of Hg2+ ions.
Experimental section
Chemicals
Deoxyribonuclease I (DNase I) and silver(I) nitrate (99+%, A.C.S. reagent) were obtained from Acros (Morris Plains, New Jersey, USA). Molecular beacon (MB) and DNA templates used to prepare DNA–Ag NCs were purchased from Integrated DNA Technology (Coralville, IA, USA). The MB used as a control (FAM-ACTTAGTTCTAAATCACTATGGTCGCACTAAGT-DABCYL) contained a carboxyfluorescein (FAM) donor and a 4-([4-(dimethylamino)phenyl]azo)benzoic acid (DABCYL). The sequences of DNA used in this study are listed in Table 1. Sodium phosphate dibasic anhydrous and sodium phosphate monobasic monohydrate (J. T. Baker, Phillipsburg, NJ, USA) were used to prepare the phosphate buffers (50 mM, pH 5.0 or 7.0). Mercury chloride, magnesium nitrate, isopropyl alcohol (IPA), 3-mercaptopropionic acid (MPA, >99.0%) and NaBH4 (powder, 98%) were purchased from Aldrich (Milwaukee, WI, USA). Milli-Q ultrapure water was used in all experiments.| Name | 1 | 2 | 3 | 4(DNATAr1) | 5(DNAAAr1) | 6 | 7 | 8 | 9 | 10(DNATAr2) | 11(DNATAr5) | 12(DNATAr6) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a λ ex: 460 nm. b λ ex: 540 nm. c —: No observed fluorescence band. d τ and Φf are the fluorescence quantum yield and lifetime of the DNA–Ag NCs, respectively. e Values of m/z obtained from ESI-MS data. f N: Number of encapsulated Ag atoms. | ||||||||||||
| Structure |
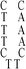
|
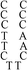
|
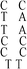
|
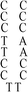
|
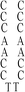
|
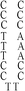
|

|
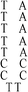
|
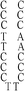
|

|
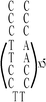
|
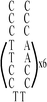
|
| λ em (nm)c | — | — | — | 536a | 546a | — | — | — | — | 608b | 620b | 644b |
| Φ f (%)d | — | — | — | 8 | 10 | — | — | — | — | 61 | 21 | 25 |
| τ (ns)d | — | — | — | — | — | — | — | — | — | 2.7 ± 0.2 | 2.6 ± 0.2 | 3.1 ± 0.3 |
| m/z (amu)e | — | — | — | 388.5 | 714.9 (domain), 730.1 (minor) | — | — | — | — | 696.0 (domain), 703.3 (minor) | 1010.6 (domain), 1019.3 (minor) | 843.7 (domain), 847.8 (minor) |
| N f | — | — | — | 2 | 2 | — | — | — | — | 4, 5 | 5, 6 | 6 |
Synthesis of DNA-Ag NCs
DNA solutions were prepared by adding the DNA stock solutions to phosphate buffer (20 mM, pH 7.0). To prepare DNA–Ag NCs, aliquots of AgNO3 solution (1 mM, 30 μL) were added separately to one of the DNA solutions (50 μM, 85 μL) to provide a Ag+-to-DNA molar ratio of 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1; this molar ratio allowed the preparation of DNA–Ag NCs at their most fluorescence.10 After incubation for 15 min in an ice bath, the mixtures were reduced by vigorously shaking with freshly prepared NaBH4 solution (2 mM, 15 μL) for approximately 30 s and then at ambient temperature in the dark for another 6 h. The as-prepared DNA–Ag NCs are named herein as DNATAr1, DNAAAr1, DNATAr2, DNATAr5, and DNATAr6–Ag NCs, according to their DNA sequences and sizes (Table 1); the subscripts 1, 2, 5, and 6 represent the number of repetitive TACC or AACC pairs in the scaffolds (e.g., TAr2 formed two TACC pairs in the presence of Ag+ ions). The concentrations of the as-prepared DNA–Ag NCs are denoted herein as 1×.
:1; this molar ratio allowed the preparation of DNA–Ag NCs at their most fluorescence.10 After incubation for 15 min in an ice bath, the mixtures were reduced by vigorously shaking with freshly prepared NaBH4 solution (2 mM, 15 μL) for approximately 30 s and then at ambient temperature in the dark for another 6 h. The as-prepared DNA–Ag NCs are named herein as DNATAr1, DNAAAr1, DNATAr2, DNATAr5, and DNATAr6–Ag NCs, according to their DNA sequences and sizes (Table 1); the subscripts 1, 2, 5, and 6 represent the number of repetitive TACC or AACC pairs in the scaffolds (e.g., TAr2 formed two TACC pairs in the presence of Ag+ ions). The concentrations of the as-prepared DNA–Ag NCs are denoted herein as 1×.
Characterization of DNA–Ag NCs
Fluorescence and absorption spectra of the as-prepared DNA–Ag NCs were recorded using a Cary Eclipse Varian spectrofluorometer (Walnut Creek, CA, USA) and a GBC Cintra 10e double-beam UV–Vis spectrophotometer (Victoria, Australia), respectively. The fluorescence lifetimes of the five distinct emitters were recorded using a photo-counting PicoHarp 300 system (PicoPicoQuant, Berlin, Germany) and a diode laser emitting at 375 nm (FluoTime 300) as the light source. Circular dichroism (CD) spectra of the DNA solution and the as-prepared DNA–Ag NCs were recorded using a JASCO 720 instrument (Easton, MD, USA).The molecular masses of the DNA strands and number of Ag atoms bound to these five distinct DNA–Ag NCs were determined using an LTQ mass spectrometer (Thermal Fisher, San Jose, CA, USA). Prior to conducting electrospray ionization mass spectrometry (ESI-MS), the as-prepared DNA–Ag NCs were purified through centrifugal filtration (13500 g, 15 min; filter cutoff: 3 kDa) and then washing with ultrapure water (3 × 450 μL) three times. Samples for ESI-MS measurement were prepared by mixing one of the DNA strand solutions or one of the DNA–Ag NC solutions (50 μL) with isopropyl alcohol (50 μL) to enhance the ionization efficiency.11 Direct ESI infusion was performed using a capillary column (375 μm o.d., 50 μm i.d.) tapered to 20 μm i.d. The flow rate was set at 250 nL min−1 using a syringe pump (New Era Pump Systems, NE-1000, Wantagh, NY); an ESI voltage of 1.8 kV was applied to the syringe for ESI infusion.
Stability of DNATAr2–Ag NCs
To evaluate the resistance of the NCs to endogenous nuclease degradation, aliquots (180 μL) of phosphate buffer (5 mM, pH 7.0) containing NaCl (10 mM), Mg(NO3)2 (5 mM), and DNATAr2–Ag NCs (0.01X) were maintained at 25 °C for 5 min prior to addition of aliquots of DNase I (final concentration: 5.0 μg mL−1). After reactions for 1 h, the mixtures were subjected to fluorescence measurement. In a control experiment, the MB (500 nM) was used in place of the DNATAr2–Ag NCs.12 To investigate thiol-induced fluorescence quenching, mixtures (200 μL) of the DNATAr2–Ag NCs (0.01×), phosphate buffer (20 mM, pH 7.0), and MPA (0–1.5 μM) were equilibrated in the dark at 27 °C for 30 min and then the mixtures were subjected to fluorescence measurement.Detection of Hg2+ ions using DNATAr2-Ag NCs
A stock HgCl2 solution (10 mM) was prepared in 1 mM HNO3. Aliquots of Hg2+ solution (0–500 nM) were diluted from the stock solution with phosphate buffer (20 mM, pH 5.0). The as-prepared DNATAr2–Ag NCs (20 μL, 0.1×) were added to the aliquots of the Hg2+ solutions to give final volumes of 200 μL. After equilibrating for 30 min at 27 °C, aliquots of the mixtures were transferred into a Synergy 4 hybrid multi-mode microplate reader (Winooski, VT, USA) for fluorescence measurement (excitation wavelength: 540 nm). Local ground water (collected from Xin-Hai tunnel, Taipei city), tap water, and drinking water were collected to evaluate the practicality of using the DNATAr2–Ag NCs as an analytical tool. Aliquots (300 μL) of the real water samples (filtered through 0.2-μm membrane) were spiked with standard Hg2+ solutions (50 μL; final concentration: 10 nM). The spiked samples were diluted to 450 μL with phosphate solution (20 mM, pH 5.0) and then analyzed using the DNATAr2–Ag NCs.Results and discussion
Effects of cytosine units on the preparation of DNA–Ag NCs
The DNA templates 1, 3, and 8, which did not contain cytosine 3-mers in the flanks of the DNA scaffolds, did not provide fluorescent products (Table 1).1a,13 The presence of cytosine 3-mers at the 5′ and 3′ ends of the DNA scaffold helped to stabilize the DNA–Ag NCs as a result of entropic effects.1cWe further investigated the effect of the distance between the repeated cytosine units (using DNA templates 4, 6, and 7) and the number of repeated cytosine units (using DNA templates 2, 4, and 9) in the DNA scaffolds. Among the DNA templates 4, 6, and 7, only the DNA template 4 led to the preparation of strongly fluorescent DNA–Ag NCs. Increasing the number of A–T pairs made it more difficult to form coordinated C–Ag–C units and, thereby, fewer fluorescent species. Comparison of the results obtained using the DNA templates 2, 4, and 9 revealed that the favorable effects of cytosine 2-mers located close to the cytosine 3-mers in the DNA scaffold; presumably, the two additional C–Ag–C bonds further stabilized the Ag NCs.7 Unexpectedly, the DNA template 9 did not form fluorescent products, suggesting that the three inside C–Ag–C bonds and the two T–A pairs destabilized the formation of the three terminal C–Ag–C units. The as-prepared DNATAr1–Ag NCs (using DNA template 4) exhibited green emission (λem = 536 nm) when excited at 460 nm, with a value of Φf of approximately 8%. Of the DNA–Ag NCs prepared from the DNA templates 4 and 5, the latter exhibited slightly stronger fluorescence (DNAAAr1–Ag NCs; Φf = ca. 10%). The two A–T pairs in the DNA template 4 presumably inhibited C–Ag–C coordination, consistent with the slightly more negative ellipticity (−5.7 mdeg) obtained for the DNATAr1–Ag NCs relative to that (−2.9 mdeg) of the DNAAAr1–Ag NCs (Table S1†).
Next, we investigated the effect of the number of repeated CC bases on the preparation of DNA–Ag NCs using the DNA templates 10–12 (Table 1). Fig. 1a reveals that the DNATAr2–, DNATAr5–, and DNATAr6–Ag NCs emitted at wavelengths of 608, 620, and 644 nm, respectively, when excited at 540 nm. Notably, the DNATAr3– and DNATAr4–Ag NCs exhibited fluorescence properties similar to those of the DNATAr2–Ag NCs (each of them has a visible absorption band near 543 nm, revealing that they have similar transition energies); as a result, we do not further discuss their performance hereafter. Fig. 1b indicates that, in addition to an absorption peak at a wavelength of 460 nm, the spectrum of the DNATAr2–Ag NCs featured a noticeable absorption band at 540 nm, resulting from a more highly discrete and quantum-confined electronic transition.3,14 Accordingly, the DNATAr2–Ag NCs exhibited stronger fluorescence (Φf = ca. 61%) when excited at 540 nm. The quantum yields (Φf) for the five DNA–Ag NCs were measured according to the literature,15 using fluorescein (Φf = 0.79) for the DNATAr1 and DNAAAr1–Ag NCs and cresyl violet (Φf = 0.54) for the DNATAr2–, DNATAr5–, DNATAr6–Ag NCs as standard fluorophores (Table 1). The maximum absorption wavelengths of the DNATAr1– and DNAAAr1–Ag NCs were 460 nm, and those of the DNATAr2–, DNATAr5–, and DNATAr6–Ag NCs were 540 nm (Fig. 1b). Each of the decaying slopes from 630–700 nm was subtracted from the absorbance of the DNA–Ag NCs. To the best of our knowledge, these Ag NCs are among the most fluorescent ever reported.1c,7a Notably, the fluorescence intensity of the DNATAr2–Ag NCs when excited at 540 nm was approximately eight-fold greater than that when excited at 460 nm. Upon increasing the number of repeated CC bases, more Ag atoms assembled in the DNA scaffolds, leading to the formation of DNA–Ag NCs of greater size. ESI-MS data (Table 1) for the DNATAr1–Ag, DNAAAr1–Ag, DNATAr2–Ag, DNATAr5–Ag, and DNATAr6–Ag NCs revealed that there were 2, 2, 4 (5 minor), 5 (6 minor), and 6 Ag atoms, respectively, in each DNA scaffold. These data support the notion that the emission wavelength underwent a red shift upon increasing the number of Ag atoms.17 The lifetimes (ca. 2–3 ns) of our DNA–Ag NCs (Table 1) were similar to those reported previously for DNA–Ag NCs.1c,7a,16
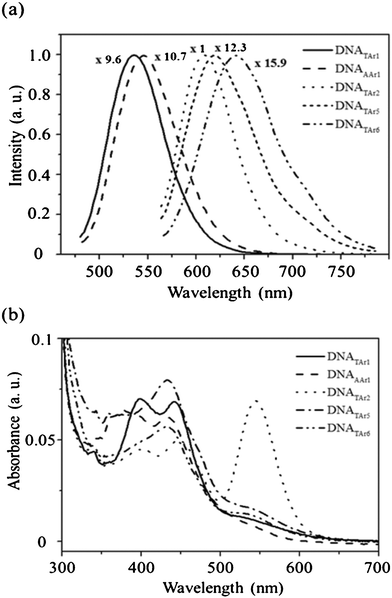 | ||
| Fig. 1 (a) Fluorescence and (b) UV–Vis spectra of the five distinct DNA–Ag NCs. Each of the mixtures, containing phosphate (20 mM, pH 7.0), AgNO3 (300 μM), a DNA template (50 μM), and NaBH4 (300 μM), was reacted at 27 °C for 6 h. The fluorescence colors of the products DNATAr1–Ag, DNAAAr1–Ag, DNATAr2–Ag, DNATAr5–Ag, and DNATAr6–Ag NCs were green, yellow, orange, saffron, and red, respectively. The fluorescence intensities have all been normalized to 1.0. Excitation wavelength: 460 nm for the first two NCs; 540 nm for the others. | ||
We employed CD to examine the changes in the DNA conformations before and after formation of the DNA–Ag NCs. The ellipticity at 284 nm of ds-DNA is typically smaller than that of ss-DNA.18 The CD data in Table S1 revealed that the hairpin-like structures of the DNA scaffolds in the DNA–Ag NCs were different from the (random-coil) structures of the DNA templates.13a The free forms of our DNA samples all exhibited positive ellipticities (mdeg)—at approximately 280 nm for DNA templates 4, 5, and 10 and at 294 nm for DNA templates 11 and 12—whereas those for the DNATAr1–, DNAAAr1–, and DNATAr2–Ag NCs were negative because the hairpin-like structures formed through C–Ag–C coordination induced the formation of nonplanar and tilted orientations of the bases relative to the helical axis.1a,10 The negative ellipticities of the DNA scaffolds in the DNA–Ag NCs were less than those in the presence of Ag+ ions (before adding NaBH4), mainly because of the weaker interactions of the DNA strands with Ag atoms than with Ag+ ions.19 The smaller changes in ellipticity for the NCs prepared from the DNA templates 11 and 12 reveal that their interactions with Ag+ ions and Ag atoms were relatively weak, mainly because of their stronger inter-DNA interactions and DNA hybridization.20 As a result, relative to the DNATAr1–, DNAAAr1–, and DNATAr2–Ag NCs, the DNATAr5– and DNATAr6–Ag NCs fluoresced more weakly.
Stability of the DNATAr2–Ag NCs
Because the DNATAr2–Ag NCs provided the highest value of Φf (61%), we expected that they could be used as a highly sensitive probe. The presence of thiols (e.g., MPA) and Cl− ions (e.g., NaCl) led to serious fluorescence quenching of DNA–Cu/Ag and DNA–Ag NCs.7a,19 Therefore, we initially investigated the effects of MPA on the fluorescence of the DNATAr2–Ag NCs.The as-prepared DNATAr2–Ag NCs were stable in the presence of MPA at concentrations of up to 0.5 μM (Fig. 2a) and their fluorescence was quenched by only 39% in the presence of 1.5 μM MPA. Thus, the as-prepared DNATAr2–Ag NCs were much more stable than our previously fabricated ones and most other reported DNA–Ag NCs, mainly because of stronger interactions between Ag atoms and the DNATAr2 scaffolds.7a,10,19 The effect of NaCl (Cl−) on the chemical stability of the DNATAr2–Ag NCs in Fig. S1.† The fluorescence intensity of the DNA–Ag NCs remained constant at the NaCl concentrations up to about 50 mM. The fluorescence intensity of the DNATAr2–Ag NCs at 4 °C remained constant for about three days and then decreased (Fig. S2†). The emission wavelength did not shift. To further support the fact that the DNA scaffold stabilized these Ag NCs, we further investigated their resistance to nuclease digestion. It is known that DNA nuclease digests the DNA templates of DNA–Ag NCs, leading to decreases in their fluorescence intensities.12,21 The fluorescence of our DNATAr2-Ag NCs decreased only by 14% in the presence of 5.0 μg mL−1 of DNA nuclease I (Fig. 2b). In the control experiment, the fluorescence of the MB was enhanced dramatically (ca. 19-fold) in the presence of 5.0 μg mL−1 of DNA nuclease I (Fig. 2c). Fluorescence resonance energy transfer occurs more efficiently when the distance between the FAM and DABCYL units is shorter (i.e., when the MB forms a hairpin structure). DNA nuclease I bound to the single-stranded loop portion of the MB and cleaved it, thereby significantly increasing the distance between the FAM and DABCYL units and restoring the fluorescence of the MB. Our results support that the DNA scaffolds were more stable on the surfaces of Ag NCs, mainly because of the formation of a Ag-mediated duplex structure.13a,b,20 The unusual stability and high value of Φf of the DNATAr2–Ag NCs in the presence of thiols, NaCl and DNA nuclease I suggest that they hold great potential for use in vivo and in vitro bioassays.
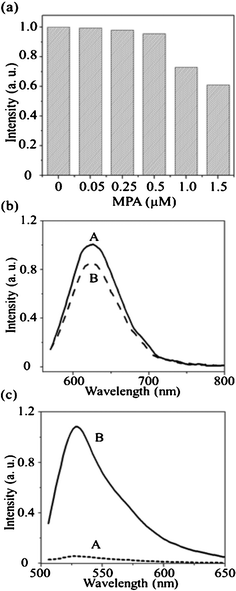 | ||
| Fig. 2 (a) Fluorescence intensities of the DNATAr2–Ag NCs (0.01×) in the presence of MPA (0, 0.05, 0.25, 0.5, 1.0, or 1.5 μM). Buffer: 20 mM phosphate (pH 7.0); incubation time: 30 min. (b, c) Fluorescence intensities of the (b) DNATAr2–Ag NCs (0.01×) and (c) MB (500 nM) in the (A) absence and (B) presence of DNase I (5.0 μg mL−1). Buffer: 5 mM phosphate buffer (pH 7.0) containing 10 mM NaCl and 5 mM Mg(NO3)2. Incubation time: 60 min. | ||
Detection of Hg2+ ions using DNATAr2–Ag NCs
Next, we employed our as-prepared DNATAr2–Ag NCs for the detection of Hg2+ ions. Monitoring of the levels of Hg2+ ions in environmental samples is an important issue because of their potentially deleterious effects on the environment and human health.22 Upon increasing the concentration (from 2.5 to 100 nM) of Hg2+ ions, the fluorescence ratio [(IF0 − IF)/IF0] of the as-prepared DNATAr2–Ag NCs increased (Fig. 3a); here, IF0 and IF represent the fluorescence intensities of the DNATAr2–Ag NCs in the absence and presence of Hg2+ ions, respectively. Recent studies have suggested that the dispersion forces between closed-shell metal atoms are highly specific and strong, particularly when they occur between Hg2+ ions (4f145d10) and silver atoms (6d10).23,24 We obtained a linear relationship (R2 = 0.98) between the value of [(IF0 − IF)/IF0] and the concentration of Hg2+ ions over the range 2.5–50 nM. The limit of detection (LOD) for Hg2+ ions (at a signal-to-noise ratio of 3) was 0.9 nM (0.18 ppb), much lower than the maximum level (2.0 ppb) permitted by the United States Environmental Protection Agency (EPA) for mercury in drinking water. This probe for Hg2+ ions provided a sensitivity that was three orders of magnitude better than those of probes featuring DNA-functionalized Au NPs.23 We further tested the selectivity of our DNATAr2–Ag probe toward Hg2+ ions (100 nM) over other common metal ions (Co2+, Fe2+, Cd2+, Zn2+, Cu2+, Mg2+, Cr3+, Mn2+, Ni2+, Pb2+, Fe3+, K+, Al3+, and Na+; each 1 μM). The selectivity values (Fig. 3b) were all greater than 80-fold; this high specificity was due mainly to strong Hg2+–Ag interactions.4c,10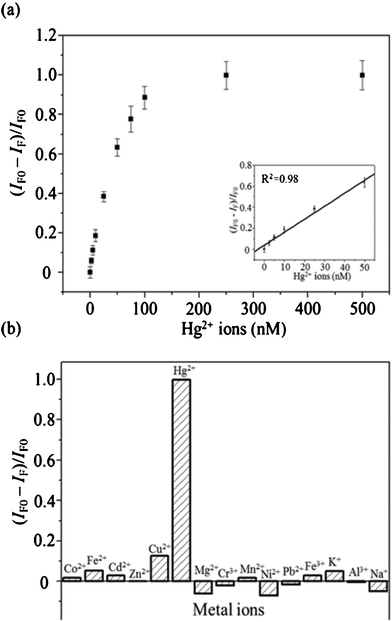 | ||
| Fig. 3 (a) Fluorescence quenching ratios plotted as a function of the concentration of Hg2+ ions. Inset: Plot of the value of (IF0 − IF)/IF0 for the DNATAr2–Ag NCs with respect to the concentration (0–50 nM) of the spiked Hg2+ ions. (b) Selectivity and sensitivity of the DNATAr2–Ag NC probe toward Hg2+ ions. Concentration of Hg2+ ions: 0.1 μM; concentrations of each other metal ion: 1 μM. DNA–Ag NCs were diluted with 20 mM phosphate solution (pH 5.0) to a final concentration of 0.004×. Reactions were performed at 25 °C for 30 min. Error bars represent standard deviations from three replicate sample measurements. | ||
We tested the practicality of our DNATAr2–Ag NC probe through the analyses of real water samples (ground water, tap water, and drinking water). We applied a standard addition curve (from 0 to 50 nM; R2 = 0.98) to determine the recoveries of Hg2+ ions (10 nM) spiked in these water samples. The high recoveries (98–118%, Table S2† in ESI) from these environmental samples reveal low matrix effects.
Conclusions
We have prepared highly fluorescent DNA–Ag NCs from AgNO3, using NaBH4 as a reducing agent, and DNA templates. ESI-MS measurements confirmed that the number of Ag atoms per DNA scaffold in the DNA–Ag NCs was highly dependent on the sequence and length of the template. Fluorescence data revealed that the number and location of the cytosine bases both played important roles in determining the optical properties of the DNA–Ag NCs. Relative to our previously prepared DNA–Ag NCs and DNA–Cu/Ag NCs, the as-prepared DNATAr2–Ag NCs in this present study were more stable and more highly fluorescent (Φf = ca. 61%).10,19 Indeed, the DNATAr2–Ag NCs provided high sensitivity and high selectivity for the detection of Hg2+ ions in water samples. Because of their biocompatibility and resistance toward MPA and nuclease digestion, these DNATAr2–Ag NCs have great potential for use in the detection of analytes in complex samples and for cell imaging.Acknowledgements
We thank the National Science Council of Taiwan for providing financial support under contracts NSC 98-2113-M-002-011-MY3, 99-2627-M-002-016, and 99-2627-M-002-017.References
- (a) J. T. Petty, J. Zheng, N. V. Hud and R. M. Dickson, J. Am. Chem. Soc., 2004, 126, 5207–5212 CrossRef CAS; (b) E. G. Gwinn, P. R. O'Neill, A. J. Guerrero, D. Bouwmeester and D. K. Fygenson, Adv. Mater., 2008, 20, 279–283 CrossRef CAS; (c) C. I. Richard, S. Choi, J.-C. Hsiang, Y. Antoku, T. Vosch, A. Bongiorno, Y.-L. Tzeng and R. M. Dickson, J. Am. Chem. Soc., 2008, 130, 5038–5039 CrossRef.
- (a) D. Takagi, Y. Homma, H. Hibino, S. Suzuki and Y. Kobayash, Nano Lett., 2006, 6, 2642–2645 CrossRef CAS; (b) J. Ramírez, M. Sanaú and E. Fernández, Angew. Chem., Int. Ed., 2008, 47, 5194–5197 CrossRef.
- (a) J. Zheng, P. R. Nicovich and R. M. Dickson, Annu. Rev. Phys. Chem., 2007, 58, 409–431 CrossRef CAS; (b) S. Link, A. Beeby, S. FitzGerald, M. A. El-Sayed, T. G. Schaaff and R. L. Whetten, J. Phys. Chem. B, 2002, 106, 3410–3415 CrossRef CAS.
- (a) C.-C. Huang, C.-K. Chiang, Z.-H. Lin, K.-H. Lee and H.-T. Chang, Anal. Chem., 2008, 80, 1497–1504 CrossRef CAS; (b) C.-C. Huang, C.-T. Chen, Y.-C. Shiang, Z.-H. Lin and H.-T. Chang, Anal. Chem., 2009, 81, 875–882 CrossRef CAS; (c) W. Guo, J. Yuan and E. Wang, Chem. Commun., 2009, 3395–3397 RSC; (d) J. Yu, S. Choi and R. M. Dickson, Angew. Chem., Int. Ed., 2009, 48, 318–320 CrossRef CAS; (e) J. Yu, S. A. Patel and R. M. Dickson, Angew. Chem., Int. Ed., 2007, 46, 2028–2030 CrossRef CAS; (f) B. Adhikari and A. Banerjee, Chem. Mater., 2010, 22, 4364–4371 CrossRef CAS; (g) Y. Antoku, J.-I. Hotta, H. Mizuno, R. M. Dickson, J. Hofkens and T. Vosch, Photochem. Photobiol. Sci., 2010, 9, 716–721 RSC.
- L. Shang and S. Dong, Chem. Commun., 2008, 1088–1090 RSC.
- (a) J. Zhang, S. Xu and E. Kumacheva, Adv. Mater., 2005, 17, 2336–2340 CrossRef CAS; (b) Z. Shen, H. Duan and H. Frey, Adv. Mater., 2007, 19, 349–352 CrossRef CAS.
- (a) J. Sharma, H.-C. Yeh, H. Yoo, J. H. Werner and J. S. Martinez, Chem. Commun., 2010, 46, 3280–3282 RSC; (b) P. R. O'Neill, L. R. Velazquez, D. G. Dunn, E. G. Gwinn and D. K. Fygenson, J. Phys. Chem. C, 2009, 113, 4229–4233 CrossRef CAS.
- H. Xu and K. S. Suslick, Adv. Mater., 2010, 22, 1078–1082 CrossRef CAS.
- T. Vosch, Y. Antoku, J.-C. Hsiang, C. I. Richards, J. I. Gonzalez and R. M. Dickson, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12616–12621 CrossRef CAS.
- G.-Y. Lan, C.-C. Huang and H.-T. Chang, Chem. Commun., 2010, 46, 1257–1259 RSC.
- T. L. Brown and J. A. Rice, Anal. Chem., 2000, 72, 384–390 CrossRef CAS.
- Y.-W. Lin, H.-T. Ho, C.-C. Huang and H.-T. Chang, Nucleic Acids Res., 2008, 36, e123 CrossRef.
- (a) C. M. Ritchie, K. R. Johnsen, J. R. Kiser, Y. Antoku, R. M. Dickson and J. T. Petty, J. Phys. Chem. C, 2007, 111, 175–181 CrossRef CAS; (b) Z. Huang, F. Pu, D. Hu, C. Wang, J. Ren and X. Qu, Chem.–Eur. J., 2011, 17, 3774–3780 CrossRef CAS; (c) S. Shukla and M. Sastry, Nanoscale, 2009, 1, 122–127 RSC.
- J. Zheng, C. Zhang and R. M. Dickson, Phys. Rev. Lett., 2004, 93, 077402 CrossRef.
- A. T. R. Williams, S. A. Winfield and J. N. Miller, Analyst, 1983, 108, 1067–1071 RSC.
- S. A. Patel, M. Cozzuol, J. M. Hales, C. I. Richards, M. Sartin, J.-C. Hsiang, T. Vosch, J. W. Perry and R. M. Dickson, J. Phys. Chem. C, 2009, 113, 20264–20270 CAS.
- (a) I. Rabin, W. Schulze, G. Ertl, C. Felix, C. Sieber, W. Harbich and J. Buttet, Chem. Phys. Lett., 2000, 320, 59–64 CrossRef CAS; (b) C. Felix, C. Sieber, W. Harbich, J. Buttet, I. Rabin, W. Schulze and G. Ertl, Chem. Phys. Lett., 1999, 313, 105–109 CrossRef CAS.
- E. Kuruvilla and D. Ramaiah, J. Phys. Chem. B, 2007, 111, 6549–6556 CrossRef CAS.
- Y.-T. Su, G.-Y. Lan, W.-Y. Chen and H.-T. Chang, Anal. Chem., 2010, 82, 8566–8572 CrossRef CAS.
- (a) N. Gerrts, T. Schmatko and E. Eiser, Langmuir, 2008, 24, 5118–5123 CrossRef; (b) F. Pierce, C. M. Sorensen and A. Chakrabarti, Langmuir, 2005, 21, 8992–8999 CrossRef CAS.
- C.-W. Liu, Y.-W. Lin, C.-C. Huang and H.-T. Chang, Biosens. Bioelectron., 2009, 24, 2541–2546 CrossRef CAS.
- (a) E. M. Nolan and S. J. Lippard, Chem. Rev., 2008, 108, 3443–3480 CrossRef CAS; (b) C.-C. Huang and H.-T. Chang, Anal. Chem., 2006, 78, 8332–8338 CrossRef CAS; (c) C.-W. Liu, Y.-T. Hsieh, C.-C. Huang, Z.-H. Lin and H.-T. Chang, Chem. Commun., 2008, 2242–2244 RSC; (d) C.-W. Liu, C.-C. Huang and H.-T. Chang, Langmuir, 2008, 24, 8346–8350 CrossRef CAS.
- (a) P. Pyykko, Angew. Chem., Int. Ed., 2004, 43, 4412–4456 CrossRef; (b) M. Kim, T. J. Taylor and F. P. Gabbai, J. Am. Chem. Soc., 2008, 130, 6332–6333 CrossRef CAS.
- J. Xie, Y. Zheng and J. Y. Ying, Chem. Commun., 2010, 46, 961–963 RSC.
Footnote |
| † Electronic Supplementary Information (ESI) available: Fig. S1–S2 and Tables S1–S2. See DOI: 10.1039/c1ra00181g/ |
| This journal is © The Royal Society of Chemistry 2011 |