Controlled hydrolysis of aryltellurium trichlorides using 2-pyrrolidinones: isolation and structural characterization of monomeric aryltellurium(IV) monohydroxides†
Shafalika
Misra
*a,
Ashok K. S.
Chauhan
*a,
Ramesh C.
Srivastava
a,
Andrew
Duthie
b and
R. J.
Butcher
c
aDepartment of Chemistry, University of Lucknow, Lucknow, 226007, India. E-mail: shafalikamisra@yahoo.co.in; akschauhan2003@yahoo.co.in
bSchool of Life and Environmental Sciences, Deakin University, Geelong, 3217, Australia
cDepartment of Chemistry, Howard University, Washington D.C. 20059, USA
First published on 30th August 2011
Abstract
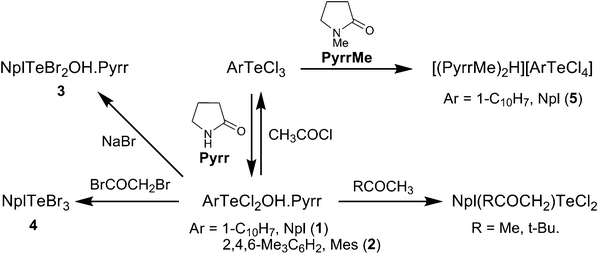
The 2-pyrrolidinone (Pyrr)-assisted hydrolysis of aryltellurium trichlorides, provided their first-step hydrolysis products as 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molecular adducts, ArTeCl2OH.Pyrr (Ar = 1-C10H7, Npl, 1; 2,4,6-Me3C6H2, Mes, 2). The hydroxo ligand occupies an equatorial position in the ψ-trigonal bipyramidal geometry around Te(IV) and is involved in a strong H-bonding interaction with the amido O of the base. As NplTeBr3 did not react with 2-pyrrolidinone under similar conditions, the bromo analogue of 1, NplTeBr2OH.Pyrr (3), was prepared by a metathetical reaction with NaBr. Both 1 and 2 undergo electrophilic substitution with acetone and pinacolone to afford Ar(RCOCH2)TeCl2 (R = Me, t-Bu), but do not react with iodoacetic acid, suggesting that the hydroxo ligand acts as a nucleophile rather than a base. The Te–OH bond in 1 is also cleaved by CH3COCl to give the parent trihalide NplTeCl3, while reaction with BrCH2COBr results in NplTeBr3 (4) as the ultimate tellurium containing product. The reaction of NplTeCl3 with N-Methyl protected 2-pyrrolidinone (PyrrMe), under similar conditions, gave an ionic complex [(PyrrMe)2H] [NplTeCl4] (5), but did not react with N-acyl protected 2-pyrrolidinone, PyrrCOCH2Br. However, the latter added oxidatively to elemental tellurium to give (PyrrCOCH2)2TeBr2 (6). In the crystal lattice of 5, discreet five-coordinate square pyramidal CTeX4 units are present, while in the case of 4 the same units are realized via intermolecular Te⋯Br interactions in a one-dimensional supramolecular architecture. N-(2-pyrrolidinone)amidomethyl ligands in 6 adopt a rare, if not the first, 1,6-(C,O) mode of chelation in preference to 1,4-(C,O) mode often observed in similar Te(IV) compounds.
:1 molecular adducts, ArTeCl2OH.Pyrr (Ar = 1-C10H7, Npl, 1; 2,4,6-Me3C6H2, Mes, 2). The hydroxo ligand occupies an equatorial position in the ψ-trigonal bipyramidal geometry around Te(IV) and is involved in a strong H-bonding interaction with the amido O of the base. As NplTeBr3 did not react with 2-pyrrolidinone under similar conditions, the bromo analogue of 1, NplTeBr2OH.Pyrr (3), was prepared by a metathetical reaction with NaBr. Both 1 and 2 undergo electrophilic substitution with acetone and pinacolone to afford Ar(RCOCH2)TeCl2 (R = Me, t-Bu), but do not react with iodoacetic acid, suggesting that the hydroxo ligand acts as a nucleophile rather than a base. The Te–OH bond in 1 is also cleaved by CH3COCl to give the parent trihalide NplTeCl3, while reaction with BrCH2COBr results in NplTeBr3 (4) as the ultimate tellurium containing product. The reaction of NplTeCl3 with N-Methyl protected 2-pyrrolidinone (PyrrMe), under similar conditions, gave an ionic complex [(PyrrMe)2H] [NplTeCl4] (5), but did not react with N-acyl protected 2-pyrrolidinone, PyrrCOCH2Br. However, the latter added oxidatively to elemental tellurium to give (PyrrCOCH2)2TeBr2 (6). In the crystal lattice of 5, discreet five-coordinate square pyramidal CTeX4 units are present, while in the case of 4 the same units are realized via intermolecular Te⋯Br interactions in a one-dimensional supramolecular architecture. N-(2-pyrrolidinone)amidomethyl ligands in 6 adopt a rare, if not the first, 1,6-(C,O) mode of chelation in preference to 1,4-(C,O) mode often observed in similar Te(IV) compounds.
Introduction
Hydrolysis of organotellurium(IV) di- and trihalides has been studied over the last century and the extent of hydrolysis is described to dependent upon the reaction conditions and electronic and steric influence of the halo and organic ligands.1 The chlorides are hydrolyzed completely in alkaline medium to afford telluroxides, R2TeO and tellurinic acids, RTe(O)OH, or the corresponding anhydrides (RTeO)2O when treated with dilute acids. Hydrolysis of aryltellurium trichlorides in neutral medium, however, affords the acid chlorides ArTe(O)Cl and seems to proceed in a stepwise manner. Monohydroxotellurium(IV) compounds, such as Ar2TeClOH or ArTeCl2OH formed in the first step of hydrolysis of Ar2TeCl2 or ArTeCl3 respectively, have not been isolated, indicating the intervening stages are fast. The isolated products of hydrolysis remained structurally ill-defined for a long time due to their low volatility and poor solubility in common organic solvents.The structure and reactivity of telluroxides and tellurinic acids has recently been the subject matter of some research groups due to their importance as potential precursors to telluroxanes, the oligomeric species that bear Te–O–M backbone linkages.2–6 This has given impetus to the development of telluroxane chemistry in analogy to the chemistry of siloxanes whose importance as molecular precursors to zeolite materials is well known.7 Precise crystallographic data that remained elusive for a long time is now known for some diaryltelluroxanes.8 The well defined dimeric tellurinic acid [2,6-Mes2C6H3Te(O)(OH)]2, stabilized kinetically with a bulky m-terphenyl ligand that limits the condensation and aggregation process,9 and monomeric [2-(C6H5N:N)C6H4Te(O)(OH)], stabilized by an intramolecular Te–N interaction,10 have recently been isolated and characterized structurally. Some novel anionic and neutral homometallic organotellurium oxide clusters bearing rings and cages (involving Te–O bonds exclusively) in their structures have also been characterized by X-ray crystallography and may be viewed as the condensation products of organotellurium trihydroxides.11,12 During the course of our study, Beckmann et al. communicated13 the crystal structure of kinetically stabilized trans-2,4,6-t-Bu3C6H2TeCl2(OH). This monohydroxide was obtained in a small amount by the solid-state hydrolysis of a secondary modification of the parent trichloride. Attempts to prepare the monohydroxide by controlled hydrolysis of the trichloride in solution failed. The hydrolysis of intramolecularly coordinated 8-Me2NC10H6TeCl3 with water afforded the dinuclear telluroxane (8-Me2NC10H6TeCl2)2O.14
In view of the hydrolytic lability of the metal and metalloid amides [M]–NR2, often obtained by aminolysis of [M]–Cl bonds,15 reactions between ArTeCl3 and 2-pyrrolidinone (γ-lactam) have been examined with the intention of initially preparing N-(aryldichlorotelluro)-pyrrolidinone, which on subsequent hydrolysis would then provide mononuclear aryltellurium(IV) hydroxides. The paramount strong hydrogen bonding tendency of γ-lactams,16 which even stabilizes pyrrolidinonium ion in the solid-state17 was expected to prevent intermolecular condensation of Te–OH bonds. This paper details (i) the preparation and structural characterization of stable aryltellurium(IV) monohydroxides and subsequent reactions involving cleavage of the Te–OH bond and (ii) the structure elucidation of products from (a) the reaction between NplTeCl3 and N-methyl-2-pyrrolidinone and (b) oxidative insertion of elemental tellurium into C–Br bond of α-bromo-N-(2-pyrrolidinone)acetamide. Also included is the X-ray crystal structure of NplTeBr3, which has surprisingly escaped elucidation so far despite those of its iodo analogue (NplTeI3)18 and other aryltellurium tribromides, viz.PhTeBr3,19 MesTeBr320 and 2-PhC6H4TeBr321 being known for a long time.
Results and discussion
Reaction of ArTeCl3 with an excess of commercial 2-pyrrolidinone at room temperature, after work-up and crystallization from dichloromethane gave the adducts, ArTeCl2OH.Pyrr (Ar = 1 − C10H7, Npl, 1; 2,4,6-Me3C6H2, Mes, 2; Pyrr = 2-pyrrolidinone) in nearly quantitative yield. The use of dried 2-pyrrolidinone resulted in a sluggish reaction with low yield. Formation of the hydroxide, ArTeCl2OH, in the first instance, appears to involve initial formation of the amide (Eqn 1a) which is hydrolyzed by the adventitious incursion of moisture present in the commercial lactam/solvent. This is substantiated by the fact that N-methylpyrrolidinone, (PyrrMe), which cannot cause aminolysis, forms an anionic complex [(PyrrMe)2H][NplTeCl4] under the same conditions. However, base assisted direct hydrolysis of the trichlorides ArTeCl3 by the moisture to give the monohydroxides, cannot be ruled out (Eqn 1b). This finds support from the fact that NplTeCl3 when stirred with water in THF affords a colorless solid, which darkens at ∼192 °C but does not melt. It is insoluble in common organic solvents and analyses for (NplTeO1.5)x. In an attempt to prepare the dihydroxide, NplTeCl(OH)2, 1 was treated separately with one mole of NaOH or 2-pyrrolidinone in water/THF. In each case the same insoluble, product was obtained. It shows that the condensation/aggregation process in the dihydroxide could not be prevented even by the H-bond formation as in case of 1.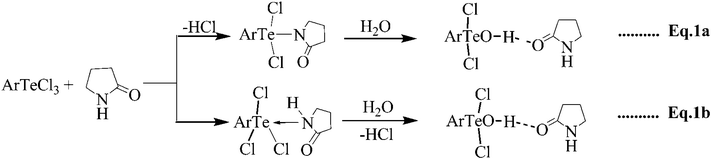
Both 1 and 2 are colorless crystalline solids soluble in dichloromethane and chloroform. They melt without decomposition, but when stirred in refluxing toluene more than 70% of adduct 1 decomposed to elemental tellurium and some insoluble, high melting solid which was not characterized as it was impossible to separate it from the Te powder completely. The chloro ligands in 1 undergo halogen exchange with NaBr to give the bromo analogue, NplTeBr2OH.Pyrr (3), which is otherwise not accessible by the reaction of NplTeBr3 with 2-pyrrolidinone. Reactions of 1 with acetone and pinacolone yield Npl(RCOCH2)TeCl2 (R = Me, t-Bu), which have been obtained earlier22 in the reaction of NplTeCl3 with the respective ketones. Formation of Npl(RCOCH2)TeCl2 from 1 would require electrophilic substitution of the ketones involving cleavage of the Te–OH bond (unless Te–Cl bond is cleaved and the liberated HCl then converts Te–OH into Te–Cl bond, which appears unlikely due to the presence of pyrrolidinone moiety). The Te–OH bond in 1 is also cleaved by acetyl chloride to afford NplTeCl3, while refluxing of 1 with α-iodoacetic acid in chloroform for 5 h left the reactants unchanged. It shows that the monohydroxide behaves as a nucleophile rather than a base. The reaction of 1 with bromoacetyl bromide is interesting as it not only cleaves the Te–OH bond but also exchanges halogens to afford NplTeBr3 instead of the mixed halogen complex NplTeCl2Br.
The N-acyl protected 2-pyrrolidinone, PyrrCOCH2Br (prepared from 2-pyrrolidinone and BrCOCH2Br), unlike PyrrMe, did not react with NplTeCl3 but adds oxidatively to elemental tellurium to give colourless crystalline (PyrrCOCH2)2TeBr2 (6).
Spectroscopic studies
Compounds 1 and 2 show νCO at ∼1635 cm−1, which is lower than that in 2-pyrrolidinone (1703 cm−1) due to its involvement in the formation of H-bonding simultaneously with the N-atom of the second molecule of pyrrolidinone and oxygen atom of the Te–OH group (vide infra). The 1H NMR spectrum of 1 consists of signals due to aryl protons between δ 7.63 and 8.60, methylene protons of the pyrrolidinone group at δ 2.16–3.42 and a broad singlet for the N–H proton. The o-Me protons as well as the two ring protons in 2 appear both as two singlets which can be explained in terms of restricted rotation along the Te–C(Mes) bond. This is also substantiated by the appearance of two signals for the o-Me carbons in the 13C NMR spectrum. The carbonyl carbon appears in the low field (δ ∼187) in 1 as well as 2 and the rest of the three carbon atoms of the pyrrolidinone group are observed as three singlets in the range δ 19–43 ppm. In compound 6, protons of methylene group bound to Te(IV) are deshielded, though marginally, compared to the parent bromide, PyrrCOCH2Br (δ 4.51). The 125Te NMR spectra of 1–6 show only a single resonance in CDCl3 suggesting the presence of only one Te containing species in solution. Interestingly the Te atom in 1 (δ 1326) is shielded compared to 2 (δ 1409) which can be attributed to greater steric requirement of mesityl group. While the electronegativity of halo ligands in NplTeCl3 and NplTeBr3 justify the observed δ125Te values (δ 1372 and 1348 respectively), the moderate shielding of Te atom in 1 compared to its bromo analogue is surprising. The δ125Te value for the anionic complex 5, as expected, is appreciably at higher field compared to that for NplTeCl3.23Molecular and crystal structures
The crystallographically determined molecular structure is unambiguous in the case of 1, while in 2, the hydroxo and one of the chloro (Cl2) ligands are two-fold disordered with occupancies 0.703:0.297. The asymmetric unit in each case comprises of a 1:1 molecular adduct of the tellurium(IV) monohydroxide with 2-pyrrolidinone, held together by polarization-assisted strong (Te)O–H⋯O(amido) hydrogen bonding interaction (Fig. 1–2). The geometry around Te(IV) in the aryltellurium(IV) monohydroxide moiety, ArTeCl2OH, of 1 and 2 resembles a distorted trigonal bipyramid with chloro ligands at the apical sites. The equatorial Te–OH bond distances of 1.884(1) Å in 1 and 1.814(4) Å in 2 are comparable to 1.993(2) Å, the value reported in case of the supermesityl analogue, trans-2,4,6-t-Bu3C6H2TeCl2OH (wherein the TeCl2OH group of atoms is two-fold disordered13). These bonds in 1 and 2 are among the shortest Te–O distances observed10 and are also shorter than the sum of covalent radii for Te and O (Σrcov(Te,O) = 2.15 Å). The greater steric demand of the mesityl ligand bound to Te(IV) in 2, is obvious from the wider equatorial C–Te–O angle of 108.80(14)° compared to 94.42(5)° in 1. Strong repulsion among the equatorial ligands and the lone pair in 2 is also evident from the exocyclic angles of 125.9(2)° and 113.7(2)° at the ipso C1 atom of the mesityl ligand that deviate from the putative value of 120°. As a consequence, local C2 symmetry of the mesityl group about the Te–C axis is lost, accounting for separate signals for the orthomethyl protons in the 1H NMR spectrum (vide supra).
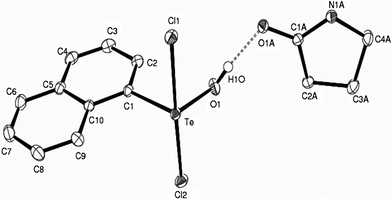 | ||
| Fig. 1 Molecular structure of 1 showing 50% probability displacement ellipsoids and the atom numbering scheme. Hydrogen atoms are omitted for clarity. Selected bond distances (Å) and angles (°): Te–C1 2.117(13), Te–Cl1 2.546(4), Te–Cl2 2.462(4), Te–O1 1.884(12), O1–H1O 0.85(3), O1–Te–C1 94.42(5), Cl1–Te–Cl2 176.68(1), Cl1–Te–C1 89.23(4), Cl1–Te–O1 89.51(4), Cl2–Te–C1 89.90 (4),Cl2–Te–O1 87.36(4). | ||
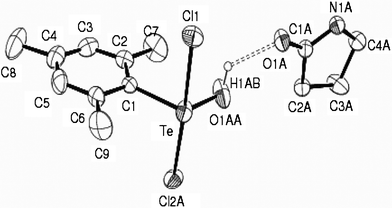 | ||
| Fig. 2 Molecular structure of 2 showing 20% probability displacement ellipsoids and the atom numbering scheme. Hydrogen atoms are omitted for clarity. Selected bond distances (Å) and angles (°): Te–C1 2.101(3), Te–Cl1 2.490(1), Te–Cl2A 2.520(1), Te–O1AA 1.814(4), O1AA–H1AB 0.82, O1AA–Te–C1 108.80(14), Cl1–Te–Cl2A 175.91(4), Cl1–Te–C1 89.05(7), Cl1–Te–O1AA 94.23(12), Cl2A–Te–C1 87.32 (7), Cl2A–Te–O1AA 85.16(12). | ||
In the lattices of compounds 1 and 2 the molecular adducts form dimers. The 2-pyrrolidinone molecules with cis amido configuration have propensity to form cyclic dimer via reciprocatory resonance-assisted N–H⋯O(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) hydrogen bonding in solution16 as well as in the metastable solid-state phase.24 Thus, dimeric structures of 1 and 2 in the crystalline state (Fig. S1 and S2, ESI†) may alternatively be described as the cyclic dimer of the lactam holding two molecules of ArTeCl2OH, one each to its amido O atoms by means of O⋯H–O hydrogen bonding interactions. The strong H-bonding interactions appear to prevent intermolecular condensation of Te–OH bonds. Each amido O atom in the centrosymmetric dimeric units of 1 and 2 holds the two protons (OH and NH) simultaneously at an angle of 120.6°, which is reminiscent of angular separation between the two lone pairs on a sp2oxygen atom. Although the eight-member ring-centered inversion point in the solid state dimeric unit of the parent lactam24 is retained in the lattices of 1 and 2, additional strong O–H⋯O hydrogen bonding interactions present in the latter bring a noticeable change in its geometry. The residual pyramidality of N atoms observed in the structure of cyclic dimer of the lactam vanishes and all its atoms (excluding C3A and C3Ai; symmetry code: i = 1.5 − x, 0.5 − y, 1 − z for 1, i = −x, −y, 1 − z for 2) become approximately coplanar (r.m.s. deviation: 0.0168 Å for 1, 0.0295 Å for 2). The dimeric lactam in adducts 1 and 2, thus adopts a nearly flat-seat chair conformation. An interesting one-dimensional supramolecular motif extending along b axis in the crystal lattice of 1 can be identified (Fig. S3†), wherein the centrosymmetric dimeric units are further associated via reciprocatory Te⋯Cl1 secondary bonding interactions of 3.550(1) Å (symmetry code: a = 1.5 − x, 1.5 − y, 1 − z). The equatorial hydroxo ligands are trans with respect to the Te2Cl2 parallelogram in the dimeric units of NplTeCl2OH (Fig. S4†) as against cis disposition, observed in the analogous dimer of 2,4,6-t-Bu3C6H2TeCl2OH.13 The role of each centrosymmetric dimeric unit of NplTeCl2OH, providing OH groups as H-bond donors in the supramolecular assembly of 1, may be compared with that of a dicarboxylic acid molecule in the lattices of the adducts of the 2-pyrrolidinone with succinic or fumaric acids.25
C) hydrogen bonding in solution16 as well as in the metastable solid-state phase.24 Thus, dimeric structures of 1 and 2 in the crystalline state (Fig. S1 and S2, ESI†) may alternatively be described as the cyclic dimer of the lactam holding two molecules of ArTeCl2OH, one each to its amido O atoms by means of O⋯H–O hydrogen bonding interactions. The strong H-bonding interactions appear to prevent intermolecular condensation of Te–OH bonds. Each amido O atom in the centrosymmetric dimeric units of 1 and 2 holds the two protons (OH and NH) simultaneously at an angle of 120.6°, which is reminiscent of angular separation between the two lone pairs on a sp2oxygen atom. Although the eight-member ring-centered inversion point in the solid state dimeric unit of the parent lactam24 is retained in the lattices of 1 and 2, additional strong O–H⋯O hydrogen bonding interactions present in the latter bring a noticeable change in its geometry. The residual pyramidality of N atoms observed in the structure of cyclic dimer of the lactam vanishes and all its atoms (excluding C3A and C3Ai; symmetry code: i = 1.5 − x, 0.5 − y, 1 − z for 1, i = −x, −y, 1 − z for 2) become approximately coplanar (r.m.s. deviation: 0.0168 Å for 1, 0.0295 Å for 2). The dimeric lactam in adducts 1 and 2, thus adopts a nearly flat-seat chair conformation. An interesting one-dimensional supramolecular motif extending along b axis in the crystal lattice of 1 can be identified (Fig. S3†), wherein the centrosymmetric dimeric units are further associated via reciprocatory Te⋯Cl1 secondary bonding interactions of 3.550(1) Å (symmetry code: a = 1.5 − x, 1.5 − y, 1 − z). The equatorial hydroxo ligands are trans with respect to the Te2Cl2 parallelogram in the dimeric units of NplTeCl2OH (Fig. S4†) as against cis disposition, observed in the analogous dimer of 2,4,6-t-Bu3C6H2TeCl2OH.13 The role of each centrosymmetric dimeric unit of NplTeCl2OH, providing OH groups as H-bond donors in the supramolecular assembly of 1, may be compared with that of a dicarboxylic acid molecule in the lattices of the adducts of the 2-pyrrolidinone with succinic or fumaric acids.25
The crystal structure of the ionic complex 5 is built of discrete tetrachloro(1-naphthyl)tellurate anions and protonated dimeric N-methyl-2-pyrrolidinone cations (Fig. 3). The five-coordinate tellurium atom in the anion [NplTeCl4]− bears slightly distorted square pyramidal geometry with mutually transoid Cl1/Cl3, Cl2/Cl4 atoms in the basal positions and ipso C1 atom of the aryl ligand in the apical position. The central Te atom lies in the basal plane (∑∠Cl–Te–Cl = 360.0°) with negligible deviation (0.0008 Å) from the mean plane of the TeCl4 unit and a narrow Te–Cl bond length range of 2.5090(4) to 2.5449(4) Å. The Te–Caryl bond length of 2.173(1) Å in 5 is slightly longer in comparison to the values reported for the analogous tetrachloroaryltellurate anions [PhTeCl4−, 2.126(2) Å26, 2.135(2) Å27; 4-PhOC6H4TeCl4−, 2.127(5) Å28 and in the zwitterion 4-H3N+-3,5-(i-Pr)2C6H2TeCl4−, 2.127(7) Å29]. It is also greater than that observed in the tetraiodo(2-naphthyl)tellurate anion (2.141(8) Å)30 or (1-naphthyl)tellurium trichloride (2.115(5) Å)23 and approaches the value for Te–Calkyl bond length, 2.178(4) Å, reported in the zwitterionic species, nacnacTeCl4.31 The Te⋯Cl and Cl⋯Cl secondary bonding interactions that are reported26 to give rise to oligomeric as well as polymeric species, [(ArTeCl4)−]n (n = 2, 3 or ∞), are absent in the crystal lattice of 5. Instead, weak intermolecular (naphthyl)C7–H7⋯Cl2 interactions self-assemble anions into chains (Fig. S5†). While centrosymmetric pairs of anions drawn from adjacent chains with face-to-face eclipsed basal planes can be identified, the internuclear distances between Te atoms [4.2335(6) Å] and eclipsed Cl atoms [either 4.1845(6) Å or 4.3293(6) Å] are too long to be of major significance. The two N-methyl-2-pyrrolidinone molecules, each of which is two-fold disordered, are held together via a strong (carbonyl)O⋯H+⋯O(carbonyl) hydrogen bond in the cation. Each N-protected molecule of the γ-lactam in the cation involves a weak (ring methylene)C2B–H2BA⋯Cl1 interaction with an anion in the neighboring one-dimensional arrays. Examples of similar protonated dimeric cations where a proton bridges (the amido O atoms unsymmetrically or ethereal O atoms symmetrically) a pair of solvent molecules by a strong hydrogen bond are available in the literature.32–34
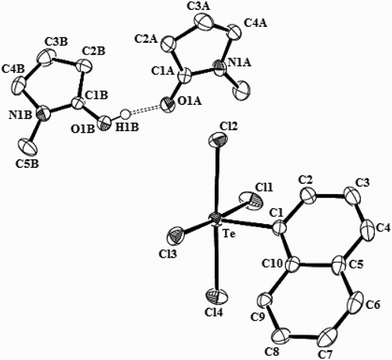 | ||
| Fig. 3 Molecular structure of 5 showing 50% probability displacement ellipsoids and the atom numbering scheme. Hydrogen atoms are omitted for clarity. Selected bond distances (Å) and angles (°): Te–C1 2.1729(14), Te–Cl1 2.5090(4), Te–Cl2 2.5298(4), Te–Cl3 2.5449(4), Te–Cl4 2.5175(4), Cl1–Te–Cl3 177.18(1), Cl2–Te–C4 177.31(1), Cl1–Te–Cl2 88.93(1), Cl2–Te–Cl3 88.70(1), Cl3–Te–Cl4 93.75(1), Cl4–Te–Cl1 88.65(1). | ||
The asymmetric unit in the crystal lattice of NplTeBr3 (4) contains two crystallographically independent molecules, each conforming to the usual ψ-trigonal bipyramidal geometry. An elliposoid diagram representing the asymmetric unit of 4 is shown in Fig. 4. While Br atom at the equatorial site is closest of the three attached to Te atom in each molecule, [d(Te1,Br12), 2.517(1)Å and d(Te2,Br23), 2.524(1)Å], the axial Te–Br bonds are unequal and substantially longer compared to Σrcov(Te,Br) value of 2.51 Å.35 Such an almost linear Te(IV)Br2 fragment among organotellurium(IV) di- and trihalides is routinely described as a three-centre four-electron bond (3c–4e), similar to that introduced by Rundle and Pimentel for trihalide anions36 and explains the hypervalency of tellurium atom without violating the octet rule or invoking ionic bonding. As two of the four electrons occupy a non-bonding molecular orbital, only one bonding pair is available for the two Te–Br bonds in 4 as well, thereby accounting for their bond orders typically less than 1.0 for a single electron pair bond (2c–2e) between the same elements. The significant disparity in the Te–Braxial bond distances in both the molecules of 4 may be due to the involvement of one of the axial Br atoms in strong intermolecular Te⋯Br secondary bonding interaction (SBI). In the crystal lattice, units of each of the independent molecules form separate polymeric zig-zag chains via μ2-bromo bridging (Fig. S6†) which are interlinked by means of Br⋯Br SBIs (Fig. S7†). The intermolecular secondary bond lengths [d(Te1⋯Br11#1) = 2.953(2)Å, d(Te2⋯Br21#2) = 2.937(2)Å] are only slightly longer (∼5%) than the longer of the two axial Te–Br bonds [d(Te1⋯Br11) = 2.800(1)Å, d(Te2⋯Br21) = 2.811(2)Å] and are shorter than Σrvdr(Te,Br) value of 4.0 Å.37 Each tellurium atom in the crystalline state of 4 thus adopts five-coordinate square pyramidal geometry with the aryl ligand at the apical site. The nearly coplanar TeBr4 {deviation from the mean plane is limited to 0.11 Å (Br12 atom) and 0.08 Å (Br23 atom) in molecules 1 and 2 respectively} occupies the basal plane. Formation of similar square pyramidal, TeX4Y units in the solid state has been observed invariably for the anions [TeX4Y]− (Y = organic ligand, X = Cl, Br or Y = X = Cl29,38) and among the oligomeric or polymeric supramolecular motifs in the solid state of similar organotellurium tribromides19 as well as organotellurium trichlorides39 including the chloro analogue of 4.23 Among the solid state structures of aryltellurium trichlorides and -tribromides, square pyramidal CTeX4 units are formed when a chloro or bromo ligand of an adjacent molecule approaches the central Te atom in a direction trans to the equatorial Cl(Br)–Te bond in preference to that of the C–Te bond resulting in two quasi linear X–Te–X alignments. Formation of such a planar covalent TeX4 moiety among 12-Te-5 hypervalent species may be said to represent a five-centre eight-electron bonding (5c–8e) system where two atomic orbitals from the Te atom and one each from four halogen atoms constitute three doubly degenerate molecular orbitals. Of the eight electrons that occupy bonding and non-bonding molecular orbitals, only four account for the four Te–Br bonds, hence weaker than a normal 2c–2e bond. Such halo-bridged square pyramidal CTeX4 units, however, are not realized in the lattices of the iodo analogue of 4,18mesityltellurium tribromide,202-biphenylyltellurium tribromide21 and also among those of the functionalized aryltellurium tribromides possessing intramolecular Te⋯A SBI with the central Te atom.40 A slight variation in the steric and electronic features of the ligands bound to the Te(IV) atom could lead to subtle modifications in the supramolecular motifs and so it is unsurprising that a diverse combination of crystal packings have been observed among organotellurium trihalides.
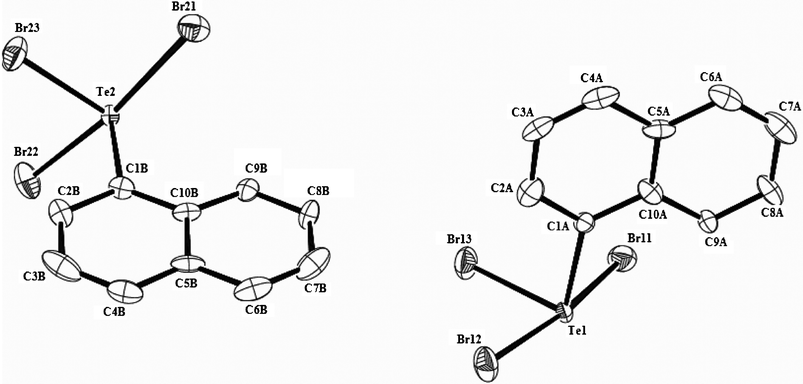 | ||
| Fig. 4 Molecular structure of 4 showing 50% probability displacement ellipsoids and the atom numbering scheme. Hydrogen atoms are omitted for clarity. Symmetry codes used to generate equivalent atoms: i = −x, 0.5 + y, 0.5 − z; ii = 1 − x, −0.5 + y, 1.5 − z. Selected bond distances (Å) and angles (°): Te1–C1A 2.130(10), Te1–Br11 2.953(2), Te1⋯Br11i 2.799(1), Te1–Br12 2.517(1), Te1–Br13 2.554(1), Te2–C1B 2.118(11), Te2–Br21 2.811(2), Te2⋯Br21ii 2.937(1), Te2–Br22 2.553(1), Te2–Br23 2.524(1); C1A–Te1–Br11 90.9(3), C1A–Te1⋯Br11i 90.4(3), C1A–Te1–Br12 96.1(3), C1A–Te1–Br13 92.5(3), Br11–Te1⋯Br11i 107.39(4), Br11–Te1–Br12 84.37(5), Br11–Te1–Br13 173.11(5), Br12–Te1⋯Br11i 84.37(5), Br12–Te1–Br13 89.12(5), Br13–Te1⋯Br11i 173.10(5), Te1⋯Br11i–Te1i 119.13(5), C1B–Te2–Br21 90.7(3), C1B–Te2⋯Br21ii 90.7(3), C1B–Te2–Br22 94.0(3), C1B–Te2–Br23 95.1(3), Br21–Te2⋯Br21ii 106.98(3), Br21–Te2–Br22 172.94(5), Br21–Te2–Br23 84.87(5), Br22–Te2⋯Br21ii 79.01(5), Br22–Te2–Br23 88.82(5), Br23–Te2⋯Br21ii 166.87(5), Te2⋯Br21ii–Te2ii 120.23(5). | ||
The molecular structure of bis(N-(2-pyrrolidinone)amidomethyl)tellurium dibromide (6), depicted in Fig. 5, presents an interesting case of intramolecular Te⋯O secondary bonding interactions. The stereochemically active lone pair at the central Te(IV) atom gives the usual ψ-trigonal bipyramidal geometry around it with bromo ligands at the apical positions. In the C-bonded organic ligands that have extended O–C–N–C–O conjugation, all the skeletal atoms are almost coplanar (r.m.s. deviations: 0.0480 Å, 0.0270 Å). The organic ligands are transoid with respect to the equatorial C–Te–C plane and subtend nearly equal angles (51.8° and 53.3°) with it resulting in an approximate C2 symmetry. The ring carbonyl O atom of each amidomethyl ligand is trans to a Te–C bond [∠O⋯Te–C = 153.97(8)°, 156.06(8)°] that makes (CO)n → σ*(Te–C) orbital interaction possible. The 1,6-(C,O) mode of chelation of the organic ligands in 6 is substantiated by the internuclear distances between the central Te and O atoms (d(Te–O2A) 2.903(2), d(Te–O2B) 2.829(2)Å) which are significantly shorter than Σrvdr(Te,O) (3.58 Å).35 Interestingly, formation of six-membered chelate rings is preferred to the four-membered alternatives, even though it results in the significant angular distortion at Csp3 atoms bound to tellurium [∠Te–Csp3–C = 114.8(2)°, 117.8(2)°]. While bis(amidomethyl)- and bis(acylmethyl)tellurium(IV) dihalides invariably exhibit 1,4-Te⋯O intramolecular attractive secondary bonding interaction,22,41 molecular structure of 6 represents one of the few (if not the only) examples of a 1,6-Te⋯O intramolecular interaction.
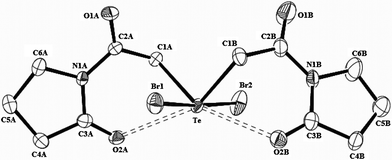 | ||
| Fig. 5 Molecular structure of 6 showing 50% probability displacement ellipsoids and the atom numbering scheme. Hydrogen atoms are omitted for clarity. Selected bond distances (Å) and angles (°): Te–C1A 2.159(2), Te–C1B 2.145(3), Te–Br1 2.6488(3), Te–Br2 2.6784(3), Te⋯O2A 2.904(2), Te⋯O2B 2.829(2); C1A–Te–C1B 93.12(11), Br1–Te–Br2 175.98(1), ClA–Te⋯O2A 64.35(8), C1B–Te⋯O2B 65.96(9), C1A–Te⋯O2B 156.06(8), C1B–Te⋯O2A 153.97(8). | ||
Experimental
General
The reactions were performed under atmospheric conditions. Solvents were dried and freshly distilled prior to use. 2-Pyrrolidinone (Merck, India) and N-Methyl-2-pyrrolidinone (Alfa-Aesar, India) were used as received. Dry 2-pyrrolidinone was obtained by storing it over CaH2 and distilling under reduced pressure. 1-Napthyltellurium trichloride and mesityltellurium trichloride were prepared by the chlorination of their corresponding ditellurides with SO2Cl2. Melting points were recorded in capillary tubes and are uncorrected. IR spectra were recorded as KBr pellets using a Perkin-Elmer RX1 spectrometer. 1H NMR spectra were recorded in CDCl3 on a Varian DRX 300 spectrometer at 300.13 MHz using Me4Si as internal standard. 13C{1H} (100.54 MHz) and 125Te{1H}(126.19MHz) NMR spectra were recorded in CDCl3 on a JEOL Eclipse Plus 400 NMR spectrometer, using Me4Si and Me2Te as internal standards. C, H, N analysis was performed on Carlo Erba model 1108 elemental analyzer. The Electrospray mass spectra were recorded on a MICROMASS QUATTRO II triple quadrupole mass spectrometer.Reactions of ArTeCl3 with 2-pyrrolidinone
:1 mixture of hexane and ethanol (3 × 3 mL). The off-white residue thus obtained was dissolved in dichloromethane (25 mL) and filtered through a short silica column. Concentration of the extract and addition of pet-ether (40°–60°) afforded a pale yellow solid which was recrystallized from chloroform to give 1 as needle shaped crystals (0.36 g, 85%); mp 120 °C. (Found: C, 39.4; H, 3.6; N, 3.1. Calc. for C14H15Cl2NO2Te: C, 39.3; H, 3.5; N, 3.2%); νmax/cm−1 1629.0 (CO). 1H NMR: δ 2.16 (2H, s, CH2), 2.35 (2H, s, CH2), 3.42 (2H, s, CH2), 6.03 (1H, s, NH), 7.63–8.6 (m, 7H, aryl). 13C{1H} NMR: δ 19.47, 28.99, 40.78 (CH2), 124.73, 125.39, 125.55, 125.73, 128.15, 130.32, 130.65, 132.94, 145.23 (Aryl C), 194.24 (CO). 125Te{1H} NMR: δ 1326; m/z (ESI) 559 [(NplTe)2O2(OH)]+, 577 [(NplTe)2O2Cl]+, 829 [(NplTe)3O4]+, 865 [(NplTe)3O3(OH)Cl]+, 883 [(NplTe)3O2(OH)3Cl]+.
When dried 2-pyrrolidinone was used in place of the commercial sample, under similar conditions, the reaction was sluggish with low yield (70%).
:1 mixture of hexane and absolute ethanol (3 × 3 mL) and dissolved in chloroform (10 mL). Addition of hexane (1 mL) and evaporation at room temperature afforded needle shaped colorless crystals of 2 (0.33 g, 80%); mp 85 °C. (Found: C, 36.76; H, 4.42; N, 3.14. Calc. for C13H19Cl2NO2Te: C, 37.19; H, 4.56; N, 3.34%); νmax/cm−1 1647.0 (CO). 1H NMR: δ 2.14 (2H, s, CH2), 2.67 (2H, s, CH2), 3.42 (2H, s, CH2), 6.38 (1H, s, NH), 2.81 (3H, s, o-Me), 2.66 (3H, s, o-Me), 2.31 (3H, s, p-Me), 6.94(1 H, s, Aryl H), 7.01 (1H, s, Aryl H). 13C{1H} NMR: δ 20.55 (p-Me), 21.04 (o-Me), 21.47 (o-Me), 19.80, 30.16, 42.62 (CH2), 130.27, 133.09, 137.41, 141.39, 142.27, 142.98 (Aryl C), 180.18 (CO). 125Te{1H} NMR: δ 1403; m/z (ESI) 543 [(MesTe)2O2(OH)]+, 561 [(MesTe)2O2Cl]+, 859 [(MesTe)3O2(OH)Cl3]+.
Npl(t-BuCOCH2)TeCl2 was obtained and characterized similarly (0.29 g, 69%); mp 168 °C (erroneously reported as 200 °C earlier22b), authentic 1H NMR.
Under similar conditions, 1 (0.42 g, 1.00 mmol) and bromoacetyl bromide (0.17 mL, 4.00 mmol) afforded orange red crystals of NplTeBr3 (4) (0.29 g, 57%); mp 156–7 °C (lit.42 159–160 °C) (Found: C, 24.3; H, 1.5. Calc. for C20H14Br6Te2: C, 24.3; H, 1.4%), 13C{1H} NMR: δ 124.79, 127.51, 127.75, 128.03, 129.66, 131.57, 133.26, 134.62 (Aryl C). 125Te{1H} NMR: δ 1348.
:1 mixture of hexane and absolute ethanol (3 × 3 mL) and dissolved in chloroform (10 mL). Addition of hexane (1 mL) and evaporation at room temperature afforded colorless chunks of 5 (0.32 g, 54%); mp 118 °C. (Found: C, 40.1; H, 4.2; N, 4.6. Calc. for C20H25Cl4N2O2Te: C, 40.4; H, 4.2; N, 4.7%); 1H NMR: δ 2.91 (3H, s, CH3), 3.69 (2H, t, CH2), 2.99 (2H, t, CH2), 2.01 (2H, t, CH2), 7.61–8.6 (m, 7H, aryl). 13C{1H} NMR: δ 17.30, 30.69, 31.14 (CH2), 51.69 (CH3), 125.53, 125.80, 126.01, 128.96, 129.47, 131.95, 132.51, 134.62, 136.92, 137.02, (Aryl C), 178.17 (CO). 125Te{1H} NMR: δ 1285.
When N-acyl-2-pyrrolidinone (PyrrCOCH2Br) was reacted with NplTeCl3, under similar conditions, the reactants were recovered unchanged.
Conclusion
First step hydrolysis products, ArTeCl2OH are isolated in good yield as hydrogen bonded adducts with 2-pyrrolidinone. However, in the absence of 2-pyrrolidinone, aqueous hydrolysis of ArTeCl3 affords (NplTeO1.5)x as an insoluble and infusible solid. The Te–OH bond in 1 is cleaved by methyl ketones and acid halides, but not iodoacetic acid. Reaction of NplTeCl3 with N-Methyl-2-pyrrolidinone yields an anionic complex [(PyrrMe)2H][NplTeCl4]. The N-acyl-2-pyrrolidinone, PyrrCOCH2Br, does not react with NplTeCl3 but adds oxidatively to elemental Te to give crystalline (PyrrCOCH2)2TeBr2 which provides the rare, if not only, example of 1,6-Te⋯O intramolecular interaction.Crystallography
Single crystals suitable for X-ray crystallography were grown from CHCl3 solutions. Intensity data were collected on Oxford Diffraction Xcalibur, Ruby and Gemini CCD diffractometers with graphite monochromated Mo-Kα (0.71073 Å). Data were reduced and corrected for absorption using spherical harmonics, implemented in SCALE3 ABSPACK scaling algorithm in the CrysAlisPro, Oxford Diffraction Ltd., version 1.171.33.55 program. The structures were solved by direct methods and difference Fourier synthesis using SHELXL-97.43 Full-matrix least-squares refinements on F2, using all data, were carried out with anisotropic displacement parameters applied to non-hydrogen atoms. Hydrogen atoms attached to carbon were included in geometrically calculated positions using a riding model and were refined isotropically. Crystallographic data collection and refinement parameters are given in Table 1. ORTEP views of molecular structures of 1, 2, 4, 5 and 6 are shown in Fig. 1–5.| 1 | 2 | 4 | 5 | 6 | |
|---|---|---|---|---|---|
| Formula | C14H15Cl2NO2Te | C13H19Cl2NO2Te | C20H14Br6Te2 | C20H25Cl4N2O2Te | C12H16Br2N2O4Te |
| Formula weight | 427.77 | 419.79 | 988.97 | 594.82 | 539.69 |
| Temperature (K) | 110(2) | 295(2) | 123(2) | 123(2) | 274(2) |
| Wavelength, λ (Å) | 0.71073 | 0.71073 | 0.71073 | 0.70173 | 0.7073 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Triclinic | Monoclinic |
| Space group | C2/c | P21/c | P21/c | P–1 | C2/c |
| a (Å) | 17.9609(5) | 13.6291(4) | 20.3174(8) | 10.0476(4) | 28.4086(10) |
| b (Å) | 7.74529(16) | 6.8780(3) | 7.3678(3) | 10.8133(3) | 9.5781(2) |
| c (Å) | 23.9193(7) | 19.3019(7) | 17.7571(8) | 11.8837(5) | 13.1481(4) |
| α | 90 | 90 | 90 | 91.270(3) | 90 |
| β | 112.645(3) | 109.336(4) | 115.656(5) | 112.651(4) | 112.596(4) |
| γ | 90 | 90 | 90 | 95.227(3) | 90 |
| V (Å3) | 3070.95(13) | 1707.33(11) | 2396.07(17) | 1184.28(8) | 3303.11(17) |
| Z | 8 | 4 | 4 | 2 | 8 |
| ρ calcd (Mg/m3) | 1.850 | 1.633 | 2.742 | 1.668 | 2.170 |
| Abs coeff. (mm−1) | 2.285 | 2.053 | 12.447 | 1.726 | 6.655 |
| F(000) | 1664 | 824 | 1793 | 590 | 2048 |
| Crystal size (mm3) | 0.48 × 0.45 × 0.34 | 0.48 × 0.41 × 0.16 | 0.87 × 0.26 × 0.16 | 0.49 × 0.38 × 0.25 | 0.49 × 0.414 × 0.37 |
| θ range (°) | 5.06 to 32.79 | 5.11 to 32.68 | 5.02 to 32.72 | 5.15 to 32.79 | 5.04 to 32.73 |
| Index ranges | −27 ≤ h ≤ 26 | −20 ≤ h≤ 20 | −29 ≤ h ≤ 24 | −14 ≤ h ≤ 15 | −43 ≤= h ≤ 40 |
| −11 ≤= k ≤ 11 | −9 ≤ k ≤ 10 | −10 ≤ k ≤ 11 | −16 ≤ k ≤ 16 | −14 ≤= k ≤ 14 | |
| −36 ≤= l ≤ 36 | −28 ≤ l ≤ 28 | −21 ≤ l ≤ 26 | −17 ≤ l ≤ 17 | −18 ≤ l ≤ 19 | |
| Reflns collected | 25706 | 13068 | 20066 | 16082 | 27863 |
| Indep reflns | 5313 [R(int) = 0.0169] | 5677 [R(int) = 0.0337] | 8027[R(int) = 0.0678] | 7808[R(int) = 0.0218] | 5736 [R(int) = 0.0516] |
| Completeness to θmax (%) | 98.9 | 98.8 | 99.1 | 99.1 | 98.7 |
| Abs. correction | Semi-empirical from equivalents | Analytical | Semi-empirical from equivalents | ||
| Max., min. transmissions | 1.00000, 0.59137 | 1.00000, 0.53504 | 0.173 , 0.026 | 1.00000 , 0.47400 | 1.00000 , 0.32999 |
| Restraints/params | 5313/0/189 | 5677/12/188 | 8027/0/254 | 7808/32/395 | 5736/0/191 |
| GoF(F2) | 1.239 | 0 .910 | 1.033 | 1.005 | 0.972 |
| Final R indices [I>2σ(I)] | R1 = 0.0189, wR2 = 0.0442 | R1 = 0.0438, wR2 = 0.1062 | R1 = 0.0686, wR2 = 0.1748 | R1 = 0.0249, wR2 = 0.0540 | R1 = 0.0325, wR2 = 0.0653 |
| R indices (all data) | R1 = 0.0202, wR2 = 0.0446 | R1 = 0.1106, wR2 = 0.1234 | R1 = 0.0963, wR2 = 0.1942 | R1 = 0.0364, wR2 = 0.0564 | R1 = 0.0535, wR2 = 0.0681 |
| Refinement method | Full-matrix least-squares on F2 | ||||
| Larg. diff. peak/hole(eÅ−3) | 0.555/−0.872 | 0.660/−0.663 | 4.068/−0.387 | 0.621/−0.649 | 1.590/−1.490 |
Acknowledgements
Financial assistance by the Department of Science and Technology, Government of India, New Delhi, is gratefully acknowledged. SM is thankful to the DST, New Delhi for Fast Track-Young Scientist Fellowship.References
- (a) K. J. Irgolic, in The Organic Chemistry of Tellurium, Gordon and Reach, New York, 1974, pp. 83, 84 and 192 and references therein Search PubMed; (b) K. J. Irgolic, J. Organomet. Chem., 1975, 103, 91 CrossRef CAS; (c) N. Petragnani, Tellurium in Organic Synthesis - Best Synthetic Methods; A. R. Katritzky, O. Meth-Cohn and C. W. Rees, Series ed.; Academic Press Limited, 1994, p. 56, 69 Search PubMed.
- M. Akiba, M. V. Lakshmikantham, K. Y. Jen and M. P. Cava, J. Organomet. Chem., 1984, 49, 4819 CAS.
- (a) S. V. Ley, C. A. Meerholz and D. H. R. Barton, Tetrahedron, 1981, 37, 213 CrossRef; (b) S. V. Ley, C. A. Meerholz and D. H. R. Barton, Tetrahedron Lett., 1980, 21, 1785 CrossRef CAS; (c) D. H. R. Barton, S. V. Ley and C. A. Meerholz, J. Chem. Soc., Chem. Commun., 1979, 755 RSC.
- (a) L. Engman, J. Lind and G. Mereenyi, J. Phys. Chem., 1994, 98, 3174 CrossRef CAS; (b) L. Engman and M. P. Cava, Tetrahedron Lett., 1981, 22, 5251 CrossRef CAS.
- (a) J. Beckmann and J. Bolsinger, Organometallics, 2007, 26, 3601 CrossRef CAS; (b) J. Beckmann, D. Dakternieks, A. Duthie, N. A. Lewcenko, C. Mitchell and M. Schürmann, Z. Anorg. Allg. Chem., 2005, 631, 1856 CrossRef CAS; (c) J. Beckmann, D. Dakternieks, A. Duthie, N. A. Lewcenko, C. Mitchell and M. Schürmann, Angew. Chem., Int. Ed., 2004, 43, 6683 CrossRef CAS; (d) J. Beckmann, D. Dakternieks, J. O'Connell, K. Jurkschat and M. Schürmann, Eur. J. Inorg. Chem., 2002, 1484 CrossRef CAS.
- (a) K. Kobayashi, K. Tanaka, H. Izawa, Y. Arai and N. Furukawa, Chem.–Eur. J., 2001, 7, 4272 CrossRef CAS; (b) K. Kobayashi, N. Deguchi, O. Takahashi, K. Tanaka, E. Horn, E. Kikuchi and N. Furukawa, Angew. Chem., Int. Ed., 1999, 38, 1638 CrossRef CAS; (c) K. Kobayashi, N. Deguchi, E. Horn and N. Furukawa, Angew. Chem., Int. Ed., 1998, 37, 984 CrossRef CAS.
- R. Murugavel, A. Voigt, M. G. Walawalkar and H. W. Röesky, Chem. Rev., 1996, 96, 2205 CrossRef CAS.
- (a) D. Naumann, W. Tyrra, R. Hermann, I. Pantenburg and M. S. Wickleder, Z. Anorg. Allg. Chem., 2002, 628, 833 CrossRef CAS; (b) T. M. Klapötke, B. Krumm, P. Mayer, H. Piotrowski and O. P. Ruscitti, Z. Naturforch., 2002, B57, 145 Search PubMed; (c) N. W. Alcock and W. D. Harrison, J. Chem. Soc., Dalton Trans., 1982, 709 RSC; (d) J. Beckmann, D. Dakternieks, A. Duthie, F. Ribot, M. Schürmann and N. A. Lewcenko, Organometallics, 2003, 22, 3257 CrossRef CAS.
- J. Beckmann, P. Finke, M. Hesse and B. Wettig, Angew. Chem., Int. Ed., 2008, 47, 9982 CrossRef CAS.
- K. Srivastava, P. Shah, H. B. Singh, U. P. Singh and R. J. Butcher, Organometallics, 2011, 30, 534 CrossRef CAS.
- H. Citeau, K. Kirschbaum, C. Conrad and D. M. Giolando, Chem. Commun., 2001, 2006 RSC.
- K. Srivastava, S. Sharma, H. B. Singh, U. P. Singh and R. J. Butcher, Chem. Commun., 2010, 46, 1130 RSC.
- J. Beckmann, S. Heitz and M. Hesse, Inorg. Chem., 2007, 46, 3275 CrossRef CAS.
- J. Beckmann, J. Bolsinger and A. Duthie, Chem. Eur. J., Int. edn, 2011, 17, 930 CAS.
- M. F. Lappert, P. P. Power, A. R. Sanger and R. C. Srivastava, Metal and Metalloid Amides, Ellis Horwood Ltd., Chichester, England, 1980 Search PubMed.
- (a) A. E. Schavlev, A. N. Pankratov, V. B. Borodulin and O. A. Chaplygina, J. Phys. Chem. A, 2005, 109, 10982 CrossRef; (b) H. Yekeler, A. Guven and R. Ozkan, J. Comput.-Aided Mol. Des., 1999, 13, 589 CrossRef CAS; (c) J. A. Walmsley, J. Phys. Chem., 1978, 82, 2031 CrossRef CAS.
- A. Bekaert, B. Viossat, J. D. Brion and P. Lemoine, Z. Kristallogr. NCS, 2009, 224, 676 CAS.
- E. S. Lang, G. M. Oliveira, E. T. Silveira, R. A. Burrow and E. M. V.-López, J. Organomet. Chem., 2002, 664, 306 CrossRef CAS.
- N. W. Alcock and W. D. Harrison, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1982, B38, 2677 CrossRef CAS.
- D. Dakternieks, J. O'Conell and E. R. T. Tiekink, J. Organomet. Chem., 2000, 598, 49 CrossRef CAS.
- C. Knobler and J. D. McCullough, Inorg. Chem., 1977, 16, 612 CrossRef CAS.
- (a) A. K. S. Chauhan, Anamika, A. Kumar, R. C. Srivastava, R. J. Butcher and A. Duthie, J. Organomet. Chem., 2006, 691, 5887 CrossRef CAS; (b) A. K. S. Chauhan, P. Singh, A. Kumar, R. C. Srivastava, R. J. Butcher and A. Duthie, Organometallics, 2007, 26, 1955 CrossRef CAS.
- A. K. S. Chauhan, unpublished work.
- R. Goddard, O. Heinemann, C. Krüger, I. Magdó, F. Mark and K. Schaffner, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1998, C54, 501 CAS.
- S. K. Callear and M. B. Hursthouse, Crystal Structure Report Archive, 2008, University of Southampton Search PubMed.
- E. S. Lang, G. M. Oliveira, R. M. Fernandes Jr. and E. M. V.-López, Z. Anorg. Allg. Chem., 2004, 630, 717 CrossRef CAS and references therein.
- S. S. Santos, E. S. Lang and R. A. Burrow, J. Braz. Chem. Soc., 2006, 17, 1566 CrossRef.
- R. K. Chadha, J. E. Drake and M. A. Khan, Can. J. Chem., 1984, 62, 32 CrossRef CAS.
- P. B. Hitchcock, M. F. Lappert and G. Li, Inorg. Chim. Acta, 2009, 362, 3982 CrossRef CAS.
- E. S. Lang, G. M. Oliveira and G. N. Ledesma, Z. Anorg. Allg. Chem., 2005, 631, 1524 CrossRef CAS.
- A. F. Gushwa, J. G. Karlin, R. A. Fleischer and A. F. Richards, J. Organomet. Chem., 2006, 691, 5069 CrossRef CAS.
- R. A. Zingaro, C. Herrera and E. A. Meyers, Phosphorus, Sulfur Silicon Relat. Elem., 1990, 48, 1 CrossRef CAS.
- S. Hasche, O. Reich, I. Beckmann and B. Krebs, Z. Anorg. Allg. Chem., 1996, 623, 724 CrossRef.
- J. Pietikäinen, A. Maaninen, R. S. Laitinen, R. Oilunkaniemi and J. Valkonen, Polyhedron, 2002, 21, 1089 CrossRef.
- L. Pauling, ‘The Nature of the Chemical Bond,’ 3rd edn, Cornell University Press, Ithaca, New York, 1960 Search PubMed.
- (a) R. J. Hatch and R. E. Rundle, J. Am. Chem. Soc., 1951, 73, 4321 CrossRef; (b) R. E. Rundle, J. Am. Chem. Soc., 1963, 85, 112 CrossRef CAS; (c) R. E. Rundle, J. Am. Chem. Soc., 1947, 69, 1327 CrossRef CAS; (d) G. C. Pimental, J. Chem. Phys., 1951, 446 CrossRef.
- J. E. Huheey in Inorganic Chemistry, 3rd ed., Harper and Row, New York, 1983, pp. 258-259 Search PubMed.
- J. Bergman, J. Siden and K. M. Moe, Tetrahedron, 1984, 40, 1607 CrossRef CAS.
- F. B. W. Einstein and T. Jones, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1982, B38, 617 CrossRef CAS.
- (a) N. Saleem, A. A. West and W. R. McWhinnie, J. Chem. Soc., Dalton Trans., 1988, 2363 RSC; (b) P. Mallikaratchy, R. E. Norman, F. R. Fronczek and T. Junk, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2003, C59, o571 CrossRef CAS.
- A. K. S. Chauhan, S. Misra, P. Singh, R. C. Srivastava, R. J. Butcher and A. Duthie, Dalton Trans., 2009, 39, 2637 Search PubMed.
- N. Petragnani, Tetrahedron, 1960, 11, 15 CrossRef.
- G. M. Sheldrick, SHELXL-97, Program for the Solution of Crystal Structures, University of Göttingen, Göttingen (Germany), 2008 Search PubMed.
- K. Brandenburg and H. Putz, Diamond (version 3.1e), Crystal Impact GbR, 2007 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The supramolecular architectures identified in the crystal lattice of 1, 2, 4 and 5 (Fig. S1–S7) using Diamond44 and H-bonding parameters in tabular form (Table S1). CCDC reference numbers for 1, 2, 4, 5 and 6796562, 796563, 825838–825840. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1ra00409c |
| This journal is © The Royal Society of Chemistry 2011 |