Poly(dialkylstannane) and poly(diarylstannane) homo- and random copolymers synthesized in liquid ammonia
Markus
Trummer
,
Debora
Solenthaler
,
Paul
Smith
and
Walter
Caseri
*
Department of Materials, Eidgenössische Technische Hochschule (ETH) Zurich, Wolfgang-Pauli-Strasse 10, CH-8093, Zurich, Switzerland. E-mail: wcaseri@mat.ethz.ch; Fax: +41 44 632 11 78; Tel: +41 44 632 22 18 Web: www.polytech.mat.ethz.ch
First published on 1st September 2011
Abstract
Random copolymers of dialkylstannanes (butyl, octyl and dodecyl) and diarylstannanes were synthesized and characterized with 119Sn NMR spectroscopy, UV/Vis spectroscopy and thermal analysis. These copolymers and the corresponding homopolymers were produced by reaction of dichlorodiorganostannanes, R2SnCl2, with sodium in liquid ammonia. In the case of dichlorodibutylstannane, Bu2SnCl2, and dichlorodiphenylstannane, Ph2SnCl2, two different reaction pathways were applied: the monomers were either directly treated with 2 molar equivalents of sodium, or reactive organostannides were formed in situ and further converted with the respective R2SnCl2. Copolymerization of Ph2SnCl2 with Oct2SnCl2 or Dod2SnCl2 did not occur by direct reaction with 2 molar equivalents of sodium, due to the low solubility of the corresponding dichlorodialkylstannanes. Therefore, synthesis of these species was conducted via the reactive phenylstannides. UV/Vis absorption spectroscopy unveiled the presence of σ-delocalization and σ-π-delocalization in the copolymers with the latter originating from the presence of SnPh2 moieties in the polymer.
1. Introduction
Polystannanes are defined as polymers of which the main chain consists of covalently connected tin atoms. This, to our knowledge, has not been reported for other metallic elements and, therefore, is of fundamental interest. Due to delocalization of electrons along the polymer backbone (σ-delocalization),1–3 polystannanes were regarded to be appealing materials with respect to their chemical, optical, thermal and electrical properties.4–10 Moreover, polystannanes with aromatic side groups were reported to feature σ-π delocalization of the electrons.11 While some research focused on copolymers of stannanes and silanes or germanes (Scheme 1),12–15 little is known about polystannane copolymers constituted of tin atoms with different organic side groups. Only a copolymer comprising of the repeat units dibutylstannane and bis(3-phenylpropyl)stannane (i.e. with the propyl and not the phenyl groups bound to the tin atoms) has been described.4 Hence, in order to further explore the spectrum of polystannanes and their materials properties, copolymers comprising dialkylstannane and diphenylstannane moieties are aimed for in this study, as indications exist that incorporation of the latter may enhance the environmental stability of these materials. Notably, poly(diphenylstannane) itself is insoluble and intractable, and these drawbacks can be circumvented by copolymerization of diphenylstannanes with dialkylstannanes.![Schematic of the reported synthesis of copolymers with diorganostannane moieties in the main chain: poly(dibutylstannane-ran-methylphenylsilane), poly(dibutylstannane-ran-dibutylsilane), poly(dibutylstannane-ran-dibutylgermane) and poly[dibutylstannane-ran-di(ω-phenylpropyl)stannane]. References indicated.](/image/article/2011/RA/c1ra00264c/c1ra00264c-s1.gif) | ||
| Scheme 1 Schematic of the reported synthesis of copolymers with diorganostannane moieties in the main chain: poly(dibutylstannane-ran-methylphenylsilane), poly(dibutylstannane-ran-dibutylsilane), poly(dibutylstannane-ran-dibutylgermane) and poly[dibutylstannane-ran-di(ω-phenylpropyl)stannane]. References indicated. | ||
Efficient dehydropolymerization of dialkylstannanes with Wilkinson's catalyst5 results in cyclic-free, linear poly(dialkylstannane)s. This route enabled exploration of materials properties of polystannanes without the influence of undesirable byproducts, in particular cyclic oligomers. Unfortunately, however, Wilkinson's catalyst was of limited applicability for polymerization of diarylstannanes, (e.g.Ph2SnH2), and according to preliminary experiments also of limited use for the synthesis of copolystannanes comprising these moieties. Therefore, we investigated if the reaction of dichlorodiorganostannanes with sodium in liquid ammonia is suited for the preparation of copolymers with diarylstannane units, by the example of poly(dialkylstannane-diphenylstannane) copolymers of the general composition (SnAlk2)x(SnPh2)y, with Alk representing an alkyl group. For reference purposes the corresponding homopolymers have also been included (Scheme 2). Conveniently, reactions of this type have found attention in the literature for some time16–19 with a focus to form tin-carbon bonds as well as tin-tin bonds,20–35 and have been investigated in more detail more recently.36,37
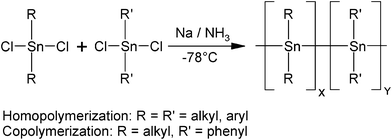 | ||
| Scheme 2 Overall reaction scheme of dichlorodialkylstannanes and dichlorodiarylstannanes with sodium for the synthesis of homo- and random copolymers in liquid ammonia. | ||
Notably, delocalization of σ-electrons in the backbone of polystannanes was found to be responsible for the characteristic yellow color and the absorption maxima around 390 nm for poly(dialkylstannane)s (σ-delocalization).5,10 In the case of aromatic side groups, additional delocalization is expected to result in a bathochromic shift of the absorption maximum, due to σ-π-delocalization.1,11 Hence, we also conducted a UV/Vis investigation with the main goal to establish if σ-π-delocalization also occurs in copolymers with phenyl and alkyl side groups.
2. Results and discussion
Polymerization
Reactions of dichlorodiorganostannanes, R2SnCl2, with sodium in liquid ammonia offer two different pathways to create polymers—either in a one-step polymerization by application of two molar equivalents of sodium (Scheme 3), or a two-step reaction of dichlorodiorganostannane with four equivalents of sodium to generate a mixture of soluble stannides as intermediates,36 followed by addition of another equivalent of dichlorodiorganostannane (two-step polymerization, Scheme 3). The two-step reaction results again in an overall dichlorodiorganostannane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Na stoichiometry of 1:2 (one sodium atom per chlorine atom), which corresponds to the theoretical ratio to yield “free (SnR2) groups” and the polymer (SnR2)n, respectively.
:Na stoichiometry of 1:2 (one sodium atom per chlorine atom), which corresponds to the theoretical ratio to yield “free (SnR2) groups” and the polymer (SnR2)n, respectively.
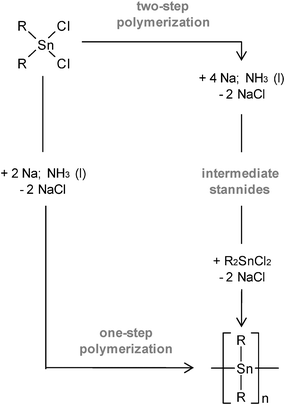 | ||
| Scheme 3 Schematic of two pathways towards poly(diorganostannane)s by reaction of dichlorodiorganostannanes with sodium in liquid ammonia. | ||
Copolymers comprising diphenylstannane and dialkylstannane units and, for comparison, the respective homopolymers are the focus of this study. A priori, it should be noted that the one- or two-step synthesis route may lead to significantly different chemical structures of the (co-)polymers, and, hence their properties may also differ. For instance, homopolymerization of dichlorodibutylstannane, Bu2SnCl2, by the two-step process is expected to result in branched polymer chains, due to the exchange of alkyl groups in the intermediate stannides.36 Yet, such phenomena have not been observed for reactions with dichlorodiphenylstannane, Ph2SnCl2, i.e. there was no evidence for exchange of aryl groups in the liquid ammonia solutions.
As indicated in the Introduction, poly(diphenylstannane) is insoluble—at least in common solvents—and increasing the length of alkyl side groups is generally reported to enhance the solubility of polymers.38–44 Hence, random copolymers of dichlorodiphenylstannane with dichlorodialkylstannanes comprising alkyl groups of various length were synthesized, i.e. with butyl (Bu), octyl (Oct) and dodecyl (Dod) groups. An overview of the homo- and copolymers explored is shown in Table 1.
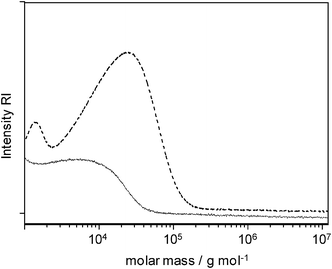 | ||
| Fig. 1 GPC traces of poly(dibutylstannane) synthesized by two-step polymerization (dashed) and poly(diphenylstannane-ran-dibutylstannane) synthesized with two-step polymerization by using Ph2SnCl2 in the first step to form intermediate diphenylstannides (solid line). | ||
| Polymer | C (% m/m) | H (% m/m) | MW (kg/mol) | PDI | ||
|---|---|---|---|---|---|---|
| found | calculated | found | calculated | |||
| (SnBu2)n | ||||||
| 1 step | 41.09 | 41.25 | 7.61 | 7.79 | 8 | 2.3 |
| 2 step | 40.75 | 7.56 | 15 | 2.2 | ||
| (SnPh2)n | ||||||
| 2 step | 51.87 | 52.81 | 3.74 | 3.69 | insoluble | — |
| (SnBu2)n(SnPh2)m | ||||||
| 1 step | 46.88 | 47.48 | 5.37 | 5.59 | 10 | 3.2 |
| 2 step (Bu-stannides) | 37.87 | 5.19 | insoluble | — | ||
| 2 step (Ph-stannides) | 47.74 | 5.50 | 10 | 3.3 | ||
| (SnOct2)n(SnPh2)m | 53.36 | 54.24 | 7.32 | 7.48 | 10 | 2.8 |
| (SnDod2)n(SnPh2)m | 57.86 | 59.21 | 8.89 | 8.28 | 12 | 3.4 |
In the two-step procedure, the stoichiometric ratios of the two Bu2SnCl2 portions of the first and the second addition were varied between 0.9 and 1.1. Isolation and subsequent dissolution of the products in THF allowed the determination of the molar mass as a function of this stoichiometry. No significant influence of the stoichiometric ratios on the molar masses was observed (Mw = 15 × 103 g/mol at a ratio of 1.1, Mw = 13 × 103 g/mol at a ratio of 0.9). Importantly, at a 10% deviation of the ratios from 1.0 a pronounced decrease in molar mass is expected for a polycondensation type reaction of the in situ formed stannides in the 1st step with dichlorodiorganostannane added in the 2nd step, according to Carother's equation.45 As this was not the case, a chain-growth polymerization is presumed to prevail, which may appear surprising at first glance as the reaction scheme may appear typical for polycondensation processes. Yet a polymerization could by initiated by radicals, as radical reactions of organostannanes in liquid ammonia are well known.29–34 The polydispersity index of 2.2 (Table 2) is in the typical range for radical chain growth polymerizations. Since the intermediates formed in situ in the first step did not react with each other to yield polymers, the species which initiated polymerization, therefore, must have been generated upon addition of the second portion of Bu2SnCl2—perhaps by reaction with sodium, as not the entire quantity of sodium is required for the formation of the observed stannides generated in the first step.36
:total stannane = 2:1, one-step synthesis). This procedure resulted in precipitation of polymeric species between 10 s and 40–50 s. The isolated polymers were largely soluble in toluene and dichloromethane (MW = 10 × 103 g/mol, Table 2, cf. also Fig. 1), but also contained an insoluble fraction. Since the corresponding homopolymers poly(dibutylstannane) and poly(diphenylstannane) differ in their solubility (see above), it would be reasonable to assume that the insoluble part consisted of copolymers of a high content of diphenylstannane. Application of mixtures of 75% mol/mol Bu2SnCl2 and 25% mol/mol Ph2SnCl2, as well as 25% mol/mol Bu2SnCl2 and 75% mol/mol Ph2SnCl2 also resulted in the formation of soluble and insoluble copolymer fractions.
The two-step copolymerization started with the in situ formation of stannides. Accordingly, there are two possibilities to produce a copolymer. If in the first step Bu2SnCl2 was applied, followed by the addition of Ph2SnCl2 in the 2nd step, the product obtained was largely insoluble in toluene and dichloromethane. Elemental analyses deviated significantly from theoretical values (Table 2). Oxygen contents of up to 5% w/w were found in the elemental compositions; obviously the materials formed were sensitive to ambient oxygen.
Employing Ph2SnCl2 in the 1st step, followed by addition of Bu2SnCl2 led to the formation of partly soluble reaction products. The soluble fraction possessed a molar mass Mw = 1 × 103 g/mol, and the elemental analyses were in good agreement with the theoretical composition (Table 2).
Thus, all copolymers yielded soluble products with Mw on the order of 104 g/mol with polydispersity indices around 3 (Table 2). The solubility facilitates characterization by NMR spectroscopy and UV/Vis spectroscopy.
Characterization
| Polymer | δ 1H (ppm) | Ratio | δ 119Sn (ppm) | ||
|---|---|---|---|---|---|
| –CH3 | –CH2– | Ar-H | Ar/Alkyl | Sn | |
| a The signal at −410 to −430 was only observed in the two-step synthesis. b Determined by solid-state MAS NMR spectroscopy. | |||||
| (SnBu2)n | 0.9–1.0 [m, 3H] | 1.1–1.8 [m, 6H] | — | — | −190, −202, −203 |
| −410 to −430a | |||||
| (SnPh2)n | — | — | — | — | −197b |
| (SnBu2)n(SnPh2)m | 1.1 [m, 3H] | 1.3–2.3 [m, 6H] | 6.7–8.1 [m, 3.7H] | 0.7–0.9 | −160 to −190 |
| −200 to −220 | |||||
| (SnOct2)n(SnPh2)m | 0.9 [t, 3H] | 1.0–1.55 [m, 14] | 6.7–7.6 [m, 5H] | 1 | −157 to −195 |
| −200 to −220 | |||||
| (SnDod2)n(SnPh2)m | 0.9 [t, 3H] | 1.0–1.65 [m, 22 H] | 6.7–7.6 [m, 5H] | 1 | −155 to −180 |
| −187 to −211 | |||||
:1 ratio between octyl and phenyl groups (Table 3). 119Sn NMR spectra displayed broad signals ranging from −157 ppm to −195 ppm resulting from SnOct2 and from −200 ppm to −220 ppm from SnPh2 moieties.
:1 ratio of dodecyl- and phenyl groups in the copolymer (Table 3, Fig. 2). 119Sn NMR spectra of the soluble polymer showed two broad peaks ranging from −155 ppm to −180 ppm, assigned to SnDod2 moieties, and from −187 ppm to −211 ppm representing SnPh2 units.
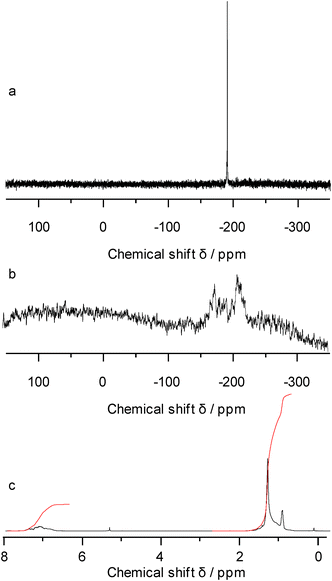 | ||
| Fig. 2 119Sn NMR spectra a) poly(didodecylstannane) and b) poly(diphenyl-ran-didodecylstannane), and c) 1H NMR spectrum of the copolymer. | ||
Note that the signals of all copolymers are broad compared to those of the poly(dialkylstannane) homopolymers (poly(diphenylstannane) is insoluble and solid-state 119Sn NMR spectra show generally broader signals). This observation is expected due to the large number of possible surroundings to each tin atom in a random copolymer. In addition, the fact that the positions of the signals of the homopolymers differ from those of the respective units in the copolymers (cf. example in Fig. 2) shows that the obtained materials do not consist of block copolymers but of random copolymers.
Homopolymers . UV/Vis absorption spectra of poly(dibutylstannane) featured an absorption maximum at 390 nm independent on the synthesis method (one- or two-step), consistent with values reported in the literature.5,6,10 Poly(diphenylstannane), however, displayed an absorption edge around 480 nm, which was attributed to enhanced delocalization of the electrons along the backbone and the aromatic side chains (σ-π-delocalization).1,11
Copolymers . UV/Vis spectra of poly(dibutylstannane-ran-diphenylstannane) synthesized by one-step reactions featured two peaks—one around 400 nm originating in the σ-delocalization of the electrons of dibutylstannane units, and one around 470 nm related to the σ-π-delocalization in diphenylstannane units. Comparing the UV/Vis spectra of the insoluble and the soluble part of poly(dibutylstannane-ran-diphenylstannane), obtained from a 1
:1 mixture of dichlorodibutylstannane and dichlorodiphenylstannane (Fig. 3a) indicated that the fraction of Ph2Sn moieties was larger in the insoluble part, as the maximum at 470 nm was more pronounced in the absorption spectra of the insoluble part (Fig. 3). The normalized spectra of the insoluble part showed an intense shoulder at a level of about 75% compared to the signal at 400 nm, whereas in the soluble part this signal was of only ∼50% of the corresponding intensity. Taking into account that the same initial molar amount of Bu2SnCl2 and Ph2SnCl2 was employed, it is obvious from the spectra that the extinction coefficient of the dibutylstannane units was higher than that of the diphenylstannane units, as absorbance at 400 nm was higher in the soluble as well as the insoluble fractions.
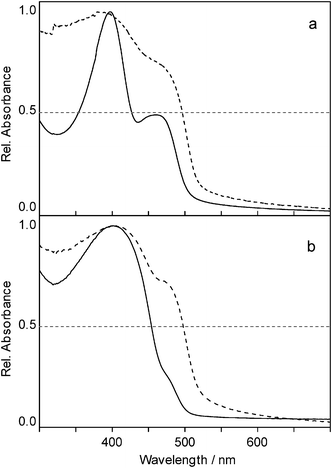 | ||
| Fig. 3 UV/Vis absorption spectra of the insoluble (dashed) and soluble (solid) fraction of poly(dibutylstannane-ran-diphenylstannane) obtained by one-step polymerization (a) and two-step polymerization (b). | ||
UV/Vis absorption spectra of the soluble polymer produced with mixtures with 25 to 75% mol/mol Bu2SnCl2 also featured the two absorption maxima at 400 nm and 470 nm. The signal at 400 nm (dibutylstannane units) increased with increasing amount of dichlorodibutylstannane in the starting mixtures concomitant with a decrease of the intensity of the signal at 470 nm, representing a decreasing fraction of diphenyltinmoieties (Fig. 4). Surprisingly, analysis of UV/Vis spectra indicated that the soluble polymer resulting from the mixture with 75% dichlorodiphenylstannane comprised a higher aromatic content than the insoluble part of the 50% mixture. Thus, it appears that not only the amount of phenyl groups, but also their arrangement had an influence on the solubility—larger segments of poly(diphenylstannane) could lead to a decreased solubility, whereas randomly distributed SnPh2 moieties, even at a higher overall diphenyltin content, can still result in solubility of the respective copolymer.
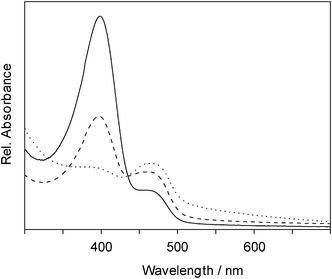 | ||
| Fig. 4 UV/Vis absorption spectra of poly(dibutylstannane-ran-diphenylstannane) produced by one-step polymerization with a molar dichlorodibutylstannane content of 75% (solid), 50% (dashed) and 25% (dotted) in the starting mixtures. | ||
A shift of the absorption maxima towards higher wavelengths was observed in the soluble part from 459 nm (25% Ph2SnCl2) to 465 nm (75% Ph2SnCl2), indicating an increasing σ-π-delocalization of the electrons in the phenyl fraction in the latter. At the same time the absorption maxima assigned to dibutylstannane units shifted from 399 nm (75% Bu2SnCl2) to 383 nm (25% Bu2SnCl2) indicative of decreasing delocalization of the (Bu2Sn) moieties (Table 4). In the case of pure poly(dibutylstannane) only σ-delocalization of the electrons occurs. The results of the above analysis represent conclusive evidence of the formation of copolymers with both diarylstannane (SnAryl2) and dialkylstannane (SnAlkyl2) moieties in the polymer main chain, as the absorption maxima shifted depending on the composition of the copolymer. This, of course would not be the case for blends of poly(dialkylstannane) and poly(diarylstannane) homopolymers. Moreover, the shifts in the absorption maxima indicate once more that the monomer units are arranged in the copolymer in a random sequence rather than in blocks.
| dibutylstannane segments | diphenylstannane segments | |||
|---|---|---|---|---|
| XButyl | EButyl | λmax | EPhenyl | λmax |
| 0.75 | 0.434 | 399 | 0.080 | 459 |
| 0.50 | 1.835 | 398 | 0.934 | 461 |
| 0.25 | 0.226 | 383 | 0.2422 | 465 |
UV/Vis absorption spectra of the materials obtained in two-step synthesis (Fig. 5b) showed a high phenyl content (assessment see above) in the insoluble part, whereas the shoulder at 470 nm in the soluble polymer, indicating the presence of diphenylstannane units, was detected, but not very pronounced.
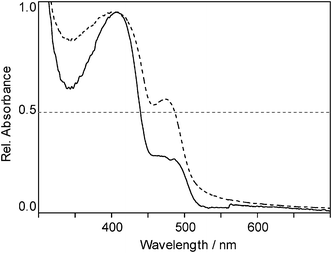 | ||
| Fig. 5 UV/Vis absorption spectra of the insoluble (dashed) and soluble (solid) fraction of poly(didodecylstannane-ran-diphenylstannane). | ||
Also the UV/Vis absorption spectra of poly(dioctylstannane-ran-diphenylstannane) featured an absorption maximum at 400 nm and a shoulder at 460 nm, representative of dioctylstannane and diphenylstannane moieties, respectively. Again, the intensity of the latter was more pronounced in the spectrum of the insoluble part.
UV/Vis absorption spectra of the soluble and insoluble parts of poly(didodecylstannane-ran-diphenylstannane) are shown in Fig. 5. Both spectra show an absorption maximum at 400 nm, together with an intense shoulder at 473 nm, which represents a bathochromic shift of the diphenylstannane related absorption maxima compared to the corresponding copolymers comprising butyl and octyl groups, respectively. The shoulder was more pronounced in the insoluble part. Furthermore, also the second absorption maximum showed a shift towards higher wavelengths when compared to the poly(dibutylstannane-ran-diphenylstannane) polymers (400 nm to 406 nm).
Overall, the UV/Vis spectra are consistent with the incorporation of aromatic moieties in the copolymer. Although there is not direct proof, the relatively random nature of the copolymers implies that the σ-π-delocalization is already established by short sequences or even individual (SnPh2) units.
Poly(dibutylstannane) synthesized with sodium in liquid ammonia did not feature the characteristic phase transition at 0 °C from a low temperature crystalline solid phase to a liquid-crystalline mesophase5,7 (Table 5). This was attributed to the presence of cyclic oligomers and the occurrence of branching points in the products obtained from liquid ammonia, due to exchange of alkyl groups,36 which would suppress crystallization of the polymer. The decomposition temperature (270 °C), however, was found to be virtually independent of the synthesis method employed; i.e. no significant differences in degradation temperatures were detected between polymers synthesized with Wilkinson's catalyst6 or sodium in liquid ammonia.
Poly(diphenylstannane) displayed no phase transition between −50 °C and 200 °C in DSC thermograms. This observation is consistent with polarization microscopy studies which revealed no change in birefringence upon heating polymer films up to 200 °C. Thermogravimetric analysis (TGA) indicated a decomposition temperature at 350 °C with an onset at about 270 °C, leading to the conclusion that poly(diphenylstannane) decomposes prior to melting.
Poly(dibutylstannane-ran-diphenylstannane) copolymers did not show any phase transition (Table 5), independent of the synthesis route (one- or two-step procedure) and the butyl/phenyl ratio (1:3 to 3:1). Also these polymers remained birefringent in the entire temperature range. Thermal decomposition occurred at 270 °C, similar to that of poly(dibutylstannane).
| Polymer | Temperature °C | Enthalpy J/g | |||
|---|---|---|---|---|---|
| 1st | 2nd | 1st | 2nd | ||
| a Referred to the synthesis method. | |||||
| (SnBu2)n | heating | −1 | — | 10.1 | — |
| Wilkinson's Cat. a | cooling | −26 | — | −9.3 | — |
| (SnBu2)n | heating | — | — | — | — |
| cooling | — | — | — | — | |
| (SnPh2)n | heating | — | — | — | — |
| cooling | — | — | — | — | |
| (SnBu2)n(SnPh2)m | heating | — | — | — | — |
| cooling | — | — | — | — | |
| (SnDod2)n(SnPh2)m | heating | 94 | — | 43.0 | — |
| cooling | 83 | — | -43.7 | — | |
| (SnDod2)n | heating | 55 | 91 | 12.7 | 2.1 |
| Wilkinson's Cat. a | cooling | 39 | 80 | -24.9 | 3.8 |
By contrast, poly(dioctylstannane-ran-diphenylstannane) showed two weak transitions in DSC thermograms at −5 °C and at 58 °C on heating (73 °C in the first heating) and at −16 °C and 52 °C upon cooling (Fig. 6). The crystallization enthalpy of the first transition was about 1.25 J/g and the second 0.65 J/g. In the corresponding homopolymer poly(dioctylstannane), two phase transitions were reported at 29 °C and 74 °C upon heating with melting enthalpies of 14.3 and 5.2 J/g, respectively.5 The decrease in the phase transition temperatures and enthalpies of the copolymers was ascribed to the lower content of octyl groups in the copolymer compared to that in the homopolymer, and the presence of rather amorphous segments of randomly dispersed diorganostannane units. Thermogravimetric analysis unveiled a slightly higher degradation temperature of 300 °C compared to the dibutylstannane-diphenylstannane copolymer.
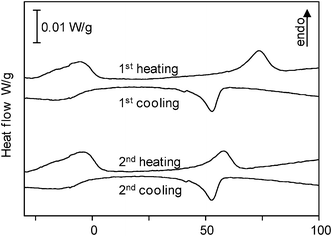 | ||
| Fig. 6 Differential scanning calorimetry (DSC) thermograms of poly(dioctylstannane-ran-diphenylstannane) recorded at heating- and cooling rates of 10 °C/min. Two phase transitions are observed at −5 °C and 58 °C upon heating and at 52 °C and −16 °C upon cooling in the second respective thermograms. | ||
Poly(didodecylstannane) homopolymer synthesized with Wilkinson's catalyst from didodecylstannane (Dod2SnH2) showed two phase transitions in DSC diagrams—at 55 °C upon heating and at 91 °C and upon cooling at 39 °C and 80 °C, with melting enthalpies of 13 J/g for the first and 2.1 J/g for the second transition. For the copolymer poly(didodecylstannane-ran-polydiphenylstannane) only one, intense transition was observed at 94 °C on heating and 83 °C on cooling with a melting enthalpy of 43 J/g (Table 5 and Fig. 7). The temperature of this transition corresponds well with the second transition of the homopolymer. It seems, therefore, that the aromatic moieties suppressed the first phase transition at 55 °C, while melting occurred at the same temperature as for the poly(didodecylstannane) homopolymer—most likely due to the long alkyl chains. Decomposition temperatures (330 °C) were found to increase compared with the corresponding homopolymers.
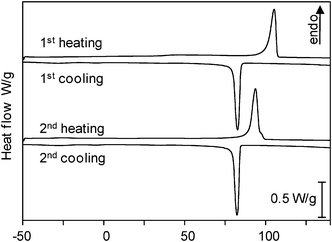 | ||
| Fig. 7 DSC thermograms of poly(didodecylstannane-ran-diphenylstannane). In contrast to poly(didodecylstannane), only one phase transition upon heating and cooling were detected. | ||
Poly(diphenylstannane), poly(dibutylstannane-ran-diphenylstannane), poly(dioctylstannane-ran-diphenylstannane) and poly(didodecylstannane-ran-diphenylstannane) were examined. Orientation was induced by shearing on a glass slide with a spatula, by drawing of solution mixed blends with UHMWPE on a hot-stage at 110 °C and, in the case of the soluble polymers, by crystallization onto PTFE orientation layers. Alignment of the polymer was analyzed by means of polarized optical microscopy, UV/Vis spectroscopy with polarized light and wide-angle X-ray diffraction (WAXD).
Shearing the polymers at room temperature induced orientation of the main chain for all materials parallel to the direction of shear, as clearly evident from Fig. 8 and 9—shown by the examples of poly(diphenylstannane) and poly(dibutylstannane-ran-diphenylstannane) (synthesized by the two-step synthesis).
 | ||
| Fig. 8 Polarized optical microscopy images taken between crossed polarizers of poly(diphenylstannane) prepared by two-step polymerization and oriented by shearing on a glass slide (top row) and by drawing of a blend with UHMWPE (second row), at 0° and 45° angles between the orientation axis of the sample and polarization direction of the light. At the bottom wide angle X-ray diffraction patterns of the oriented materials are shown; left, oriented by shearing; right, by drawing of blends with UHMWPE. Arrows indicate signals attributed to the polystannane, whereas the other signals originate from UHMWPE; orientation direction vertical. | ||
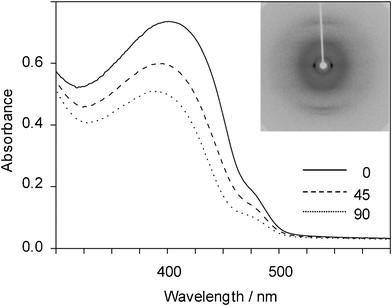 | ||
| Fig. 9 Polarized UV/Vis absorption spectra (angles relative to the orientation direction indicated) and wide angle X-ray diffraction pattern of poly(dibutylstannane-ran-diphenylstannane) oriented by shearing (orientation direction vertical). | ||
Tensile deformation of blends with UHMWPE provided the same results for poly(diphenylstannane), poly(dibutylstannane-ran-diphenylstannane) and poly(dioctylstannane-ran-diphenylstannane). The orientation of these polymers was parallel to the drawing direction. Drawing of blends with poly(didodecylstannane-ran-diphenylstannane), on the other hand, showed unexpected results. UV/Vis spectroscopy with polarized light yielded different results compared to those recorded in the shearing experiments. The development of the shoulder at 470 nm, attributed to the aromatic content of the copolymer, with the angle between polarization direction of incident light and drawing direction was essentially the same for both methods (Fig. 10). By contrast, that of the signal around 400 nm differed strikingly (cf.Fig. 10b). Dichroism at 400 nm was more pronounced and the absorbance perpendicular to the drawing direction and polarization plane of incident light was higher, i.e. the dichroic ratio changed from 1.2 to −1.7 (the negative sign indicates that absorption perpendicular to the polarization direction is higher than parallel). This result implies that the copolymerization of dichlorodiphenylstannane and dichlorodidodecylstannane did not yield an alternating copolymer, but contains didodecylstannane segments which cause orientation of side groups parallel to the drawing axis thus forcing orientation of the corresponding main chain segment to be perpendicular to the drawing axis, as was observed with poly(didodecylstannane) homopolymer.5
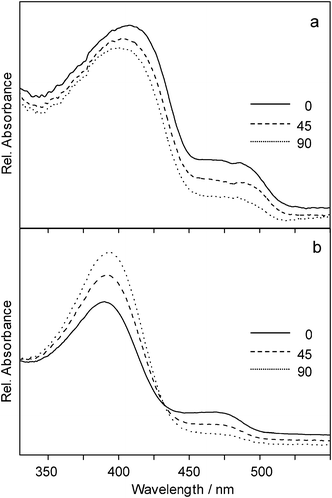 | ||
| Fig. 10 Polarized optical absorption spectra of poly(didodecylstannane-ran-diphenylstannane) oriented by shearing on a glass slide at room temperature (a) and by drawing of a blend with UHMWPE (b). Note that for orientation by shearing the absorption at 0° is higher for both signals, whereas in the drawing experiments the absorption around 400 nm is higher at 90°. | ||
Inducing orientation by crystallization from solution onto pre-oriented PTFE layer was not possible for poly(diphenylstannane) due to its insolubility and was not successful for the copolymers.
3. Conclusions
We demonstrated that formation of polystannane homopolymers and copolymers with sodium in liquid ammonia can be achieved via two different reaction paths. That is, starting from dichlorodiorganostannanes, R2SnCl2, one-step synthesis by reaction with two molar equivalents sodium and two-step synthesis, where in the first step reactive intermediates are generated by the reaction with four molar equivalents sodium. The two-step polymerization does not follow a step-growth mechanism, but rather chain growth polymerization, probably initiated by radicals generated during the second synthetic step. Migration of alkyl groups–but not of phenyl groups–during the reaction with sodium in liquid ammonia was confirmed, which can lead to branched polymers if alkylstannide intermediates are formed.By means of 119Sn NMR spectroscopy, UV/Vis spectroscopy and thermal analyses it is demonstrated that copolymers were created and not blends of the respective homopolymers. UV/Vis spectra indicated the presence of both σ-π delocalization and pure σ-delocalization in poly(dialkylstannane-ran-diphenylstannane).
Phase transitions observed in homopolymers were not, or only partially reflected in the related copolymers. The decomposition temperatures of poly(dialkylstannane-ran-diphenylstannane)s increased with increasing length of the alkyl group.
Like the homopolymers, the copolymers could readily be oriented by shearing or by drawing of blends with ultra-high molecular weight polyethylene (UHMWPE). In most cases, the polystannane main chains oriented preferentially parallel to the orientation direction of the external stimulus. However, interestingly, the orientation of poly(didodecylstannane-ran-diphenylstannane) depended on the orientation method. Whilst in simple shearing experiment the copolymer chain axis oriented into the direction of shear, this polymer revealed preferential orientation of segments with dodecylstannane perpendicular and segments with diphenylstannane parallel to the drawing direction of blends with UHMWPE.
4. Experimental
Materials
Ammonia was purchased from PanGas (Dagmarsellen, Switzerland, 99.999%). Dichlorodibutylstannane and dichlorodioctylstannane were acquired from ABCR GmbH (Karlsruhe, Germany) and dichlorodiphenylstannane from Sigma Aldrich (Buchs, Switzerland). The substances were recrystallized twice by dissolving in boiling pentane and subsequent precipitation at −78 °C. Deuterated dichloromethane CD2Cl2 (99.9% D) was purchased from Cambridge Isotope Laboratories (ReseaChem GmbH, Burgdorf, Switzerland), and organic solvents from Fluka (Buchs, Switzerland). Dichlorodidodecylstannane was synthesized according to literature.5Characterization
NMR spectra were recorded on a BrukerUltraShield 300 MHz/54 mm Fourier transform spectrometer with standard 5 mm broad band probe. All soluble samples were dissolved in CD2Cl2. Solid state magic-angle spinning experiments were executed on a BrukerUltraShield 500 MHz/54 mm Fourier transform spectrometer, at a spinning rate of 14 000 rpm. Samples were prepared in BrukerPh MAS ZrO2 Rotors with Kel-F caps.Elemental analyses were performed by the Microelemental Analysis Laboratory of the Department of Chemistry at ETH Zürich. Gel permeation chromatography was conducted with a GPC instrument from Viscotek (VE7510) equipped with degasser, VE1121 solvent pump, VE520 autosampler and Model 301 triple detector array. A PL gel 5 μm Mixed-D column from Polymer Laboratories Ltd. (Shropshire, United Kingdom) calibrated with atactic poly(styrene) standards from Fluka (Buchs, Switzerland) were used. Samples were dissolved in THF with 2.5% v/v toluene which served as a marker. THF eluent flow amounted to 1 mL/min. For optical microscopy, a Leica DMRX microscope equipped with two polarizers and a Mettler Toledo FP82 HT hot stage was used. UV/Vis spectra were recorded in transmission with a Perkin Elmer Lambda 900 spectrophotometer equipped with rotating polarizers. The insoluble materials were measured on quartz glass slides as films prepared by mechanical shearing, the soluble products were dissolved in tetrahydrofuran. Thermal analysis were carried out with a differential scanning calorimeter (DSC) DSC822e instrument (Mettler Toledo, Greifensee, Switzerland) equipped with an intracooler, and thermal gravimetric analysis (TGA) with a TGA/SDTA851e from Mettler Toledo under nitrogen atmosphere. The heating and cooling rates were 10 °C/min Maximum decomposition was determined by the maximum of the 1st derivative of the TGA thermogram. Wide-angle X-ray diffraction pattern were taken with a Diffraction Xcalibur™ PX (Oxford Instruments, Scotts Valley, USA), using Mo Kα radiation.
Polymerization
:1) mixture until no chloride was detected in the washing solution (usually 3–4 times, until the addition of 5 mL saturated AgNO3 solution did not lead to the formation of visible AgCl precipitates) and again dried in vacuum (0.1 mbar, 24 h).
| # steps | Na (mmol) | 1st step (mmol) | 2nd step (mmol) | |
|---|---|---|---|---|
| Homopolymers | ||||
| Poly(dibutylstannane) | 1 | 15.21 | 7.610 Bu | |
| 2 | 3.72 | 1.860 Bu | 1.865 Bu | |
| Poly(diphenylstannane) | 1 | 13.24 | 6.625 Ph | |
| 2 | 19.42 | 4.850 Ph | 4.852 Ph | |
| Copolymers | ||||
| Poly(dibutylstannane -ran- diphenylstannane) | 1 | 14.58 | 3.645 Bu + 3.64 Ph | |
| 2 | 15.11 | 3.780 Bu | 3.775 Ph | |
| 2 | 16.94 | 4.242 Ph | 4.239 Bu | |
| Poly(dioctylstannane -ran- diphenylstannane) | 2 | 14.84 | 3.710 Ph | 3.710 Oct |
| Poly(didodecylstannane -ran- diphenylstannane) | 2 | 19.88 | 4.970 Ph | 4.968 Dod |
:1) mixture until no chloride was detected in the washing solution (as before, usually 3–4 times, until the addition of 5 mL saturated AgNO3 solution did not lead to the formation of AgCl precipitates) and again dried in vacuum (0.1 mbar, 24 h).
References
- R. D. Miller and J. Michl, Chem. Rev., 1989, 89, 1359–1410 CrossRef CAS
.
- L. R. Sita, Organometallics, 1992, 11, 1442–1444 CrossRef CAS
- L. R. Sita, K. W. Terry and K. Shibata, J. Am. Chem. Soc., 1995, 117, 8049–8050 CrossRef CAS
- F. Choffat, Y. Buchmüller, C. Mensing, P. Smith and W. Caseri, J. Inorg. Organomet. Polym. Mater., 2009, 19, 166–175 CrossRef CAS
- F. Choffat, D. Schmid, W. Caseri, P. Wolfer and P. Smith, Macromolecules, 2007, 40, 7878–7889 CrossRef CAS
- F. Choffat, P. Smith and W. Caseri, J. Mater. Chem., 2005, 15, 1789–1792 RSC
- M. P. de Haas, F. Choffat, W. Caseri, P. Smith and J. M. Warman, Adv. Mater., 2006, 18, 44–47 CrossRef CAS
- F. Choffat, P. Smith and W. Caseri, Adv. Mater., 2008, 20, 2225–2229 CrossRef CAS
- F. Choffat, P. Wolfer, P. Smith and W. Caseri, Macromol. Mater. Eng., 2010, 295, 210–221 CrossRef CAS
- F. Choffat, S. Fornera, P. Smith, W. R. Caseri, D. W. Breiby, J. W. Andreasen and M. M. Nielsen, Adv. Funct. Mater., 2008, 18, 2301–2308 CrossRef CAS
- V. Y. Lu and T. D. Tilley, Macromolecules, 2000, 33, 2403–2412 CrossRef CAS
- S. J. Holder, R. G. Jones, R. E. Benfield and M. J. Went, Polymer, 1996, 37, 3477–3479 CrossRef CAS
- A. Mustafa, M. Achilleos, J. Ruiz-Iban, J. Davies, R. E. Benfield, R. G. Jones, D. Grandjean and S. J. Holder, React. Funct. Polym., 2006, 66, 123–135 CrossRef CAS
- M. Okano, N. Matsumoto, M. Arakawa, T. Tsuruta and H. Hamano, Chem. Commun., 1998, 1799–1800 RSC
- M. Okano and K. Watanabe, Electrochem. Commun., 2000, 2, 471–474 CrossRef CAS
- R. F. Chambers and P. C. Scherer, J. Am. Chem. Soc., 1926, 48, 1054–1062 CrossRef CAS
- C. A. Kraus and W. V. Sessions, J. Am. Chem. Soc., 1925, 47, 2361–2368 CrossRef CAS
- R. H. Bullard and W. B. Robinson, J. Am. Chem. Soc., 1927, 49, 1368–1373 CrossRef CAS
- C. A. Kraus and W. N. Greer, J. Am. Chem. Soc., 1925, 47, 2568–2575 CrossRef CAS
- T. Harada, Sci. Papers Inst. Phys. Chem. Res., 1939, 35, 302–313 Search PubMed
- T. Harada, J. Sci. Res. Inst. Tokyo, 1940, 38, 146–166 CAS
- T. Harada, J. Sci. Res. Inst. Tokyo, 1948, 43, 31–33 CAS
-
R. K. Ingham and H. Gilman, Inorganic Polymers, Academic Press, New York Search PubMed
- R. K. Ingham, S. D. Rosenberg and H. Gilman, Chem. Rev., 1960, 60, 459–539 CrossRef CAS
- K. Kühlein, W. P. Neumann and H. Mohring, Angew. Chem., Int. Ed. Engl., 1968, 7, 455–456 CrossRef
- M. Mąkosza and K. Grela, Synth. Commun., 1998, 28, 2697–2702 CrossRef
- M. F. Connil, B. Jousseaume, N. Noiret and M. Pereyre, Organometallics, 1994, 13, 24–25 CrossRef CAS
- C. Beermann and C. Hartmann, Z. Anorg. Allg. Chem., 1954, 276, 20–32 CrossRef CAS
- E. F. Córsico and R. A. Rossi, J. Org. Chem., 2002, 67, 3311–3316 CrossRef
- E. F. Córsico and R. A. Rossi, Synlett, 2000, 227–229 CrossRef
- C. C. Yammal, J. C. Podesta and R. A. Rossi, J. Org. Chem., 1992, 57, 5720–5725 CrossRef CAS
- A. B. Chopa, M. T. Lockhart and V. B. Dorn, Organometallics, 2002, 21, 1425–1429 CrossRef CAS
- A. B. Chopa, M. T. Lockhart and G. Silbestri, Organometallics, 2001, 20, 3358–3360 CrossRef CAS
- V. B. Dorn, M. A. Badajoz, M. T. Lockhart, A. B. Chopa and A. B. Pierini, J. Organomet. Chem., 2008, 693, 2458–2462 CrossRef CAS
- K. R. Wursthorn, H. G. Kuivila and G. F. Smith, J. Am. Chem. Soc., 1978, 100, 2779–2789 CrossRef CAS
- M. Trummer and W. Caseri, Organometallics, 2010, 29, 3862–3867 CrossRef CAS
- M. Trummer, J. Zemp, C. Sax, P. Smith and W. Caseri, J. Organomet. Chem., 2011, 696, 3041–3049 CrossRef CAS
- J. Majnusz, J. M. Catala and R. W. Lenz, Eur. Polym. J., 1983, 19, 1043–1046 CrossRef CAS
- H.-R. Dicke and R. W. Lenz, J. Polym. Sci., Polym. Chem. Ed., 1983, 21, 2581–2588 CrossRef CAS
- G. V. Laivins and D. G. Gray, Macromolecules, 1985, 18, 1746–1752 CrossRef CAS
- M. Ballauff, Macromolecules, 1986, 19, 1366–1374 CrossRef CAS
- M. Ballauff, Makromol. Chem. Rapid Commun., 1986, 7, 407–414 CrossRef CAS
- M. Ballauff, Angew. Chem., 1989, 101, 261–276 CrossRef CAS
- W. R. Krigbaum, H. Hakemi and R. Kotek, Macromolecules, 1985, 18, 965–973 CrossRef CAS
- W. H. Carothers, Trans. Faraday Soc., 1936, 32, 39–49 RSC
- T. N. Mitchell and M. El-Behairy, J. Organomet. Chem., 1977, 141, 43–48 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2011 |