Highly fluorescent organic nanoparticles of thiacyanine dye: A synergetic effect of intermolecular H-aggregation and restricted intramolecular rotation†
Hiroshi
Yao
* and
Koji
Ashiba
Graduate School of Material Science, University of Hyogo, 3-2-1 Koto, Kamigori-cho, Ako-gun, Hyogo, 678-1297, Japan. E-mail: yao@sci.u-hyogo.ac.jp; Fax: 81 791 58 0161; Tel: 81 791 58 0160
First published on 1st September 2011
Abstract
Highly fluorescent organic nanoparticles are synthesized via ion-association between 3,3′-diethylthiacyanine (TC) cation and tetrakis(4-fluorophenyl)borate anion in the presence of a neutral stabilizing polymer in water. The fluorescence shows a significant increase with a decrease in the nanoparticle size, resulting in a high fluorescence quantum yield of about 0.8–0.9. The observed fluorescence enhancement is due to a synergetic effect of both restriction of intramolecular rotation of TC and intermolecular H-aggregation in an inclined manner.
Introduction
Recently discovered fluorescent organic nanoparticles (FONs) are expected to play roles in a wide variety of applications such as nano-sized organic light emitting diodes (OLEDs) and biologics.1,2 In general, many organic materials are highly emissive in their dilute solutions but become weakly luminescent when fabricated into solids (that is, aggregation-caused quenching) mostly due to intermolecular energy transfer and/or fast internal conversion processes,3,4 so to mitigate the quenching and to obtain high quality FONs, several chemical/physical approaches have been developed; for example, phenyl-substituted siloles5 and 1-cyano-trans-1,2-bis(4′-methylbiphenyl)ethylene,3a which are virtually nonemissive in solutions but their solid films are highly luminescent, are synthesized for the nanoparticle constituents. On this basis, a concept of aggregation-induced enhanced emission (AIEE) has been proposed, which is caused by the restriction (or immobilization) of intramolecular rotation of fluorophores.5a Meanwhile, we have developed a new route for the synthesis organic nanoparticles in aqueous solution based on the ion-association technique that has advantages in simplicity and versatility.6 This method utilizes the formation of water-insoluble ion-pair aggregates in aqueous phases by association of a chromophoric ion with a hydrophobic counterion to fabricate organic nanoarchitectures. Using this approach, we are able to immobilize chromophores (fluorophores) monomolecularly in the particle.In this paper, we report the ion-based synthesis of highly fluorescent organic dye nanoparticles with various particle sizes. We find that observed intense fluorescence originates from a combined effect of restricted “intramolecular” rotation and “intermolecular” H-aggregation of the dye molecules. Much emphasis is placed on the fact that nanoparticle’s fluorescence increases with a decrease in the particle size that can be tuned by changing the molar ratio between the dye and hydrophobic counterion. 3,3′-Diethylthiacyanine is selected as the cationic dye (fluorophore) because radiationless deactivation of this dye has been accounted for simply in terms of rotational relaxation to an intermediate funnel state on the singlet potential surface, followed by undergoing radiationless decay toward the ground states;7 hence an increase in the media viscosity, which can be achieved by ion-based nanoparticle formation, slows down such molecular motions, resulting in a reduction of non-radiative deactivation and in an increase in the fluorescence quantum yield of this dye.8 Interestingly, formation of an H-type aggregate, possessing a well-defined hypsochromic shift with respect to the monomer resonance, gives an additional and significant contribution to the fluorescence enhancement for the organic dye nanoparticles. A great influence of H-aggregation on the fluorescence emission is particularly remarkable in consideration with the fact that classical H-aggregates are generally non- or weakly fluorescent.3b
Experimental
Materials
3,3′-Diethylthiacyanine (TC; chemical structure is shown in Fig. 1a) chloride was purchased from Hayashibara Biochemical Laboratories and used as received. Poly(vinylpyrrolidone) (PVP; average MW = 10 000, Aldrich) was used as a neutral stabilizer to prevent particle agglomeration. Sodium tetrakis(4-fluorophenyl)borate dihydrate (Na–TFPB•2H2O, Aldrich; see also Fig. 1a) was of the highest commercial grade available and used as received without further purification. Pure water was obtained by an Advantec GS-200 automatic water-distillation supplier.![(a) Absorption spectra of native TC (TC–Cl) in water and in chloroform. Negative solvatochromism can be seen. (b) Absorption spectra of TC nanoparticles prepared at the molar ratio ρ of 1, 2, and 4. The molar ratio is defined as ρ = [TFPB]/[TC]. The inset displays subtraction that was calculated from absorption spectra of nanoparticles and solution-phase (chloroform) with normalized intensities at the monomer peak position.](/image/article/2011/RA/c1ra00497b/c1ra00497b-f1.gif) | ||
| Fig. 1 (a) Absorption spectra of native TC (TC–Cl) in water and in chloroform. Negative solvatochromism can be seen. (b) Absorption spectra of TC nanoparticles prepared at the molar ratio ρ of 1, 2, and 4. The molar ratio is defined as ρ = [TFPB]/[TC]. The inset displays subtraction that was calculated from absorption spectra of nanoparticles and solution-phase (chloroform) with normalized intensities at the monomer peak position. | ||
Instrumentation
Morphology and size of the nanoparticles were examined with a Hitachi S-4800 scanning transmission electron microscope (STEM). A specimen for STEM observations was prepared by dropping the suspension on an amorphous carbon-coated copper mesh. The hydrodynamic diameter measurements of nanoparticles on the basis of dynamic light scattering (DLS) were conducted with an Otsuka ELS-800 light scattering spectrophotometer with a 10-mW He–Ne laser. The crystallinity of the solid-state precipitates was examined with a polarized-light microscope (BXP; Olympus) with cross polarizers since it gives us significant information on the structural anisotropy with birefringence. UV-visible absorption spectra were recorded on a Hitachi U-4100 spectrophotometer. Fluorescence spectra were obtained with a Hitachi F-4500 spectrofluorometer.Fluorescence quantum yields (Φf) were determined by comparing the emission spectra of TC in water (Φf = 2.0 × 10−3) obtained with excitation at 407 nm.8a In the measurements, we set the absorbance of 407 nm at around 0.1–0.15 and calculated Φf values with corrections for the absorbances of all dispersions.
Synthesis
Results and discussion
We first measured absorption spectra of TC–Cl in bulk water and in chloroform (Fig. 1a). The absorption maximum of TC monomer varied from 421 nm (water, dielectric constant ε = 78.4) to 430 nm (chloroform, ε = 4.8), representing negative solvatochromism; that is, the absorption is shifted to blue in more polar solvents.8a In addition, the spectrum of TC strongly depended on the dye concentration in aqueous solution;9 it is a well-known phenomenon that the intensity of monomer peak becomes weaker with increasing the dye concentration and a second peak observed at higher energy (403 nm), attributed to an H-type aggregate, stronger.9 Note that we here use the term H-type aggregate, which includes also dimers, on the basis of the hypsochromic spectral shift of the absorption peak relative to the respective monomer band.A series of TC nanoparticles dispersed in water were successfully synthesized, which was confirmed by their characteristic Tyndall scattering. Fig. 1b shows typical absorption spectra of TC nanoparticle samples prepared at ρ = 1, 2 and 4. In comparison with the spectrum of TC–Cl in aqueous solution, the monomer peak was red-shifted to around 430 nm for the nanoparticle samples. The most significant feature is the observation of a prominent second peak centered at higher energy of 407 nm. This peak can be attributed to an H-type aggregate of TC.9 Interestingly, with an increase in ρ, the absorbance ratio of A407/A430, where the suffix represents the monitor wavelength, also increased; A407/A430 = 1.26, 1.46, and 1.50 for ρ = 1, 2, and 4, respectively. The fact suggests that large ρ values facilitate the formation of H-type aggregates. The red shift in the monomer peak position stems from the solvent (in this case, TFPB “matrix”) effect as has been observed for the solution-phase TC chromophore.6,9
Fig. 2 shows typical results on DLS data along with STEM images of the TC nanoparticles prepared at ρ = 1, 2, and 4. According to the DLS data, the mean particle diameter decreased with an increase in ρ, that is, 135.2 nm at ρ = 1, 51.6 nm at ρ = 2, and 20.5 nm at ρ = 4. On the other hand, the STEM images reveal that the samples have the particle diameters in the range of 40–90 nm (ρ = 1), 30–60 nm (ρ = 2), and 15–40 nm (ρ = 4). In nanoparticles prepared at ρ = 2 and 4, the sizes determined by DLS accord well with those estimated by STEM, whereas the DLS data for the nanoparticle sample prepared at ρ = 1 appear to show the size of the agglomerates of TC nanoparticles with their diameter of 40–90 nm. In any case, the particle diameter decreases with an increase in ρ. This behavior is quite similar to that observed for pseudoisocyanine (PIC) nanoparticles synthesized in the same ion-based manner.6a In the cyanine nanoparticles consisting of PIC cations and TFPB anions, it has been revealed that the size decrease with increasing ρ originates from excess adsorption of TFPB anions onto the nanoparticle surfaces, which would suppress the growth of particles.6 The increase in surface adsorption of TFPB can bring about the reduction of the surface tension of nanoparticles according to the Gibbs’ adsorption equation,10,6 resulting in the decrease in particle size. Note that similar size-variation behaviors are also observed for metal oxide nanoparticles.11
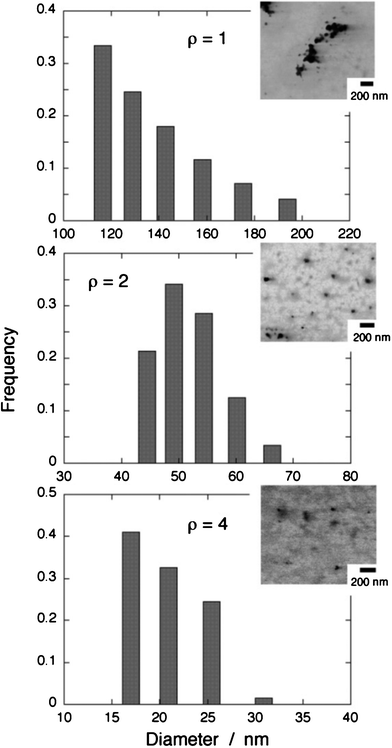 | ||
| Fig. 2 Size distributions of the TC nanoparticle samples characterized by DLS. The corresponding STEM images are also shown. | ||
To examine the composition of the solid-phase nanoparticle, precipitates formed by the ion-association reaction at ρ = 1 were collected using centrifugation and analyzed. Note that no PVP was added to make the solid-state precipitates grow to be large in size. The IR spectrum of the purified solid product had characteristic features of both TC and TFPB. Hence we can conclude that the solid-phase TC particles consist of ion-pair aggregates of TC cations and TFPB anions. It is worth noting that the ion-pair solid was revealed to be amorphous by polarized-light microscopy. See the Supplementary Information for more details.†
Surprisingly, we observed a greatly enhanced fluorescence from TC nanoparticles. A series of fluorescence spectra of the TC nanoparticles prepared at ρ = 1, 2, and 4 are shown in Fig. 3a, together with the fluorescence of native TC–Cl in aqueous solution (inset). Note that the samples with absorption properties shown in Fig. 1b were excited at 407 nm (higher energy peak). Fluorescence of aqueous TC solution was very weak, but on formation of nanoparticlesvia ion-association, a substantially enhanced intensity of fluorescence was found. The photo in the inset in Fig. 3b taken under 365 nm UV light irradiation showed the evidence of strong emission from TC nanoparticles. Of particular interest is that as the nanoparticles become smaller, or, as the ρ value increases, the fluorescence becomes more intense. For example, from Fig. 3a, fluorescence of TC nanoparticles prepared at ρ = 4 has the intensity with 220-fold enhancement compared to that of TC–Cl in water. Note that this enhancement factor is just an approximation since the absorbance correction has not been conducted here.
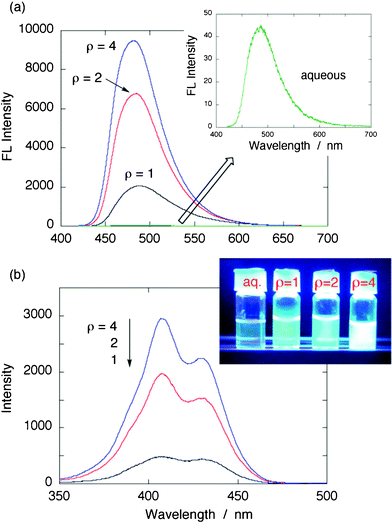 | ||
| Fig. 3 (a) Fluorescence spectra of TC nanoparticle samples prepared at ρ = 1, 2, and 4, along with that of TC–Cl in aqueous solution (λex = 407 nm). The inset shows magnified fluorescence spectrum of TC in aqueous solution. The corresponding absorption spectra are displayed in Fig. 1. (b) Excitation spectra of TC nanoparticle samples. The fluorescence was monitored at 480 nm. For the measurements, samples were diluted to avoid spectral distortion. The inset shows a photo of fluorescence taken under 365 nm UV light irradiation (original samples). | ||
To more quantitatively estimate the fluorescence quantum yields (Φf) of TC nanoparticles, all samples were diluted with water appropriately. The fluorescence of TC in aqueous solution was used as the reference.8a Then the Φf values of the TC nanoparticles were obtained as follows; 0.07–0.10 (ρ = 1), 0.40–0.48 (ρ = 2), and 0.76–0.88 (ρ = 4). The emission efficiency was approaching close to unity. This observed phenomenon of enhanced fluorescence of nanoparticles resembles that of monomeric TC in viscous media such as glycerol or in bacterial cells; for example, Φf = 0.32 for glycerol.8a It is known that intramolecular rotational motions in TC provide pathways for non-radiative decay,7,9 so an increase in the media viscosity can slow down such molecular motions, resulting in the increase in the fluorescence quantum yield of TC. In the ion-based nanoparticles, TC molecules are immobilized in the system and thus its molecular motions or rotations are restricted, yielding an significant increase in the dye fluorescence.
One should recall that the increase in the TC fluorescence intensity is strongly associated with the increase in the amount of H-type aggregates relative to that of monomer. Therefore, in addition to the effect of restriction of intramolecular rotation, fluorescence from the H-type aggregate should also contribute to its enhancement.‡ This is confirmed by the excitation spectra of TC nanoparticles (Fig. 3b).6c From Fig. 3b, the excitation spectrum was quite similar to the absorption spectrum for every sample. This intermolecular effect arises from interactions between π molecules of TC and depends strongly on their packing structure.3b In general, according to an exciton theory,3b H-aggregates tend to accelerate nonradiative relaxation because of strong π-stacking interactions in a parallel (face-to-face) aligned structure. Although our observation seems to contradict the common perception that H-type aggregates are not fluorescent, it still complies with the exciton coupling theory.3b,9 For simplicity, two molecules are considered here. If these molecules are slantingly parallel with respect to each other, the interaction of two molecular transition moments yields an energetic splitting of the excited state into two components that are allowed. In such a case, two absorption bands appear in the spectrum. The difference between the absorption energies is known as the Davydov splitting energy.9 To check the Davydov splitting in our TC nanoparticle system, we calculated the H-type aggregate spectrum by subtracting the monomer spectrum from the overall nanoparticle spectrum. Note that an absorption intensity of almost zero was assumed for the H-type aggregate at the wavelength of the monomer maximum based on the exciton theory,3b,9 and we chose the monomer spectrum as that observed in chloroform (with the same peak position). The inset in Fig. 1b (green curve) shows the obtained spectrum of the H-type aggregate in the TC nanoparticles. As expected, the presence of detectable absorption band at 443 nm results in a non-vanishing transition probability between the ground and the lowest exciton state from which a fluorescence arises.9 Hence the observed fluorescence enhancement for TC nanoparticles can be attributed both to the intramolecular rotational restriction and intermolecular H-type aggregation with inclined geometry, that is, a synergetic effect. Mostly, aggregate formation in a small aggregation number (typically dimer) is practical, so we next discuss why the H-type dimer geometry of TC molecules is preferable in the presence of (stoichiometric or excess) TFPB anions using quantum chemical calculations.
We made simple structural models of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 or 2:3 ion-pair complexes of TC:TFPB to examine how stoichiometric or excess counteranions in the vicinity of TC dimers influence their geometry. The 2:3 complex corresponds to the model of TC nanoparticles prepared in a condition using excess TFPB anions, that is, at a large ρ value. We optimized the ground-state geometry of TC2–TFPB2 or (TC2–TFPB3)− complexes with the Gaussian 09 program at the semi-empirical PM6 level.12 We considered two different initial configurations for each complex, one starting from two stacked TC molecules surrounded by two or three TFPB anions, and the other starting from two separated TC molecules surrounded by two or three TFPB anions. Fig. 4 (left) displays the optimized structures of the 2:2 and 2:3 complexes of TC and TFPB. Their relative stability is also deduced based on the total energy. The right panel in Fig. 4 shows the side views of two TC molecules extracted from the corresponding complexes. Our simplified model provided relatively long intermolecular distances of about 5.7 Å in the TC dimers, but this can be a good indication for evaluating stable configurations. The calculations suggest that an H-type stacking geometry is less stable compared to the monomeric (separated) configuration in the 2:2 complexes (Fig. 4a and 4b), whereas the situation is altered in the 2:3 complexes; the H-type stacking of TC molecules in an inclined manner is more preferable than the monomeric geometry. Therefore, it is reasonable to conclude that excess TFPB, which corresponds to a large ρ value condition (or situation that the excess TFPB can adsorb onto the nanoparticle surface), allows to facilitate the formation of inclined H-type aggregates including dimer, which can be fluorescent. We believe that TC nanoparticles are a new class of FONs whose emission originates from both restricted intramolecular rotation and intermolecular H-aggregation of the dye molecules, and will have a number of potential applications in nanosized devices.
:2 or 2:3 ion-pair complexes of TC:TFPB to examine how stoichiometric or excess counteranions in the vicinity of TC dimers influence their geometry. The 2:3 complex corresponds to the model of TC nanoparticles prepared in a condition using excess TFPB anions, that is, at a large ρ value. We optimized the ground-state geometry of TC2–TFPB2 or (TC2–TFPB3)− complexes with the Gaussian 09 program at the semi-empirical PM6 level.12 We considered two different initial configurations for each complex, one starting from two stacked TC molecules surrounded by two or three TFPB anions, and the other starting from two separated TC molecules surrounded by two or three TFPB anions. Fig. 4 (left) displays the optimized structures of the 2:2 and 2:3 complexes of TC and TFPB. Their relative stability is also deduced based on the total energy. The right panel in Fig. 4 shows the side views of two TC molecules extracted from the corresponding complexes. Our simplified model provided relatively long intermolecular distances of about 5.7 Å in the TC dimers, but this can be a good indication for evaluating stable configurations. The calculations suggest that an H-type stacking geometry is less stable compared to the monomeric (separated) configuration in the 2:2 complexes (Fig. 4a and 4b), whereas the situation is altered in the 2:3 complexes; the H-type stacking of TC molecules in an inclined manner is more preferable than the monomeric geometry. Therefore, it is reasonable to conclude that excess TFPB, which corresponds to a large ρ value condition (or situation that the excess TFPB can adsorb onto the nanoparticle surface), allows to facilitate the formation of inclined H-type aggregates including dimer, which can be fluorescent. We believe that TC nanoparticles are a new class of FONs whose emission originates from both restricted intramolecular rotation and intermolecular H-aggregation of the dye molecules, and will have a number of potential applications in nanosized devices.
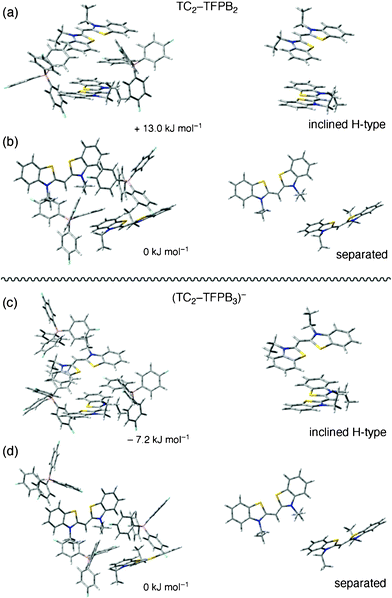 | ||
| Fig. 4 (a) and (b): Optimized structures of 2:2 ion-pair complexes of TC2–TFPB2 calculated at the PM6 level. (c) And (d): Optimized structures of 2:3 ion-pair complexes of TC2–TFPB3 calculated at the PM6 level. The right-side images show sole TC geometries extracted from the complexes shown in the left-side images. The energy value expresses the calculated relative stability with respect to the more stable geometry shown in Fig. 4b and 4d, in which two TC chromophores are distributed separately. | ||
Conclusions
In summary, highly fluorescent organic nanoparticles of thiacyanine dye were synthesized by the ion-association method. Size tuning of amorphous TC nanoparticles could be conducted by varying the molar ratio of the loaded anion to the dye cation. In comparison with TC monomer in water, the nanoparticles showed a significant increase in their fluorescence. The fluorescence intensity increased with a decrease in the nanoparticle size, resulting in a very high quantum yield of about 0.8–0.9. The fluorescence enhancement can be attributed to the synergetic effect of restriction of intramolecular rotation of TC and intermolecular H-type aggregation in an inclined manner. The simple and versatile synthesis techniques based on ion-association will provide improvement in the fluorescence properties in a new type of organic nanoparticles.Acknowledgements
The present work was financially supported by Grant-in-Aids for Scientific Research (C: 22510104 (H. Y.)) from Japan Society for the Promotion of Science (JSPS).References
- (a) H. Kasai, H. Kamatani, S. Okada, H. Oikawa, H. Matsuda and H. Nakanishi, Jpn. J. Appl. Phys., 1996, 4, L221 Search PubMed; (b) H. Kasai, H. Kamatani, Y. Yoshikawa, S. Okada, H. Oikawa, A. Watanabe, O. Itoh and H. Nakanishi, Chem. Lett., 1997, 1181 CrossRef CAS.
- (a) H.-B. Fu and J.-N. Yao, J. Am. Chem. Soc., 2001, 123, 1434 CrossRef CAS; (b) S. J. Lim, B. K. An, S. D. Jung, M. A. Chung and S. Y. Park, Angew. Chem., Int. Ed., 2004, 43, 6346 CrossRef CAS; (c) D. Horn and J. Rieger, Angew. Chem., Int. Ed., 2001, 40, 4330 CrossRef CAS.
- (a) B.-K. An, S.-K. Kwon, S.-D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410 CrossRef CAS; (b) M. Kasha, H. R. Rawls and M. A. El-Bayoumi, Pure Appl. Chem., 1965, 11, 371 CrossRef CAS.
- R. H. Friend, R. W. Gymer, A. B. Holmes, J. H. Burroughes, R. N. Marks, C. Taliani, D. D. C. Bradley, D. A. Dos Santos, J. L. Bredas, M. Logdlund and W. R. Salaneck, Nature, 1999, 397, 121 CrossRef CAS.
- (a) J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740 RSC; (b) J. Chen, C. C. W. Law, J. W. Y. Lam, Y. Dong, S. M. F. Lo, I. D. Williams, D. Zhu and B. Z. Tang, Chem. Mater., 2003, 15, 1535 CrossRef CAS; (c) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332 RSC.
- (a) H. Yao, Z. Ou and K. Kimura, Chem. Lett., 2005, 34, 1108 CrossRef CAS; (b) Z. Ou, H. Yao and K. Kimura, Bull. Chem. Soc. Jpn., 2007, 80, 295 CrossRef CAS; (c) H. Yao, M. Yamashita and K. Kimura, Langmuir, 2009, 25, 1131 CrossRef CAS.
- M. R. V. Sahyun and J. T. Blair, J. Photochem. Photobiol., A, 1997, 104, 179 CrossRef CAS.
- (a) M. S. Thomas, V. Nunez, S. Upadhyayula, E. R. Zielins, D. Bao, J. M. Vasquez, B. Bahmani and V. I. Vullev, Langmuir, 2010, 26, 9756 CrossRef CAS; (b) A.-Y. Jee, H. Kwon and M. Lee, J. Chem. Phys., 2009, 131, 171104 CrossRef.
- (a) C. Peyratout and L. Daehne, Phys. Chem. Chem. Phys., 2002, 4, 3032 RSC; (b) U. Rösch, S. Yao, R. Wortmann and F. Würtner, Angew. Chem., Int. Ed., 2006, 45, 7026 CrossRef.
- B. G. J. Moody, R. K. Owusu, A. M. Z. Slawin, N. Spencer, J. F. Stoddart, J. D. R. Thomas and D. J. Williams, Angew. Chem., Int. Ed. Engl., 1987, 26, 890 CrossRef.
- (a) L. Vayssieres, A. Hagfeldt and S. E. Lindquist, Pure Appl. Chem., 2000, 72, 47 CrossRef CAS; (b) L. Vayssieres, N. Beermann, S.-E. Lindquist and A. Hagfeldt, Chem. Mater., 2001, 13, 233 CrossRef CAS.
- (a) J. J. P. Stewart, J. Mol. Model., 2007, 13, 1173 CrossRef CAS; (b) M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. BaroneB. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. FoxGaussian 09, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
Footnotes |
| † Electronic Supplementary Information (ESI) available: FT-IR spectrum of the solid-state product of TC–TFPB along with those of pure Na–TFPB and TC–Cl, and polarized-light microscopy observations of the solid-state product. See DOI: 10.1039/c1ra00497b |
| ‡ Emission from a specific complex of TC and TFPB may be possible. However, it can be ruled out because it is well known that the thiacyanine-based specific 1:1 ion-pair (strong) complex has a fluorescence peak centered at longer wavelength than that of monomer, whereas the H-aggregate has a similar fluorescence spectrum to that of monomer (ref. 9a). |
| This journal is © The Royal Society of Chemistry 2011 |