Synthesis of PbS nanocrystals from sulfur–amine solutions at room temperature†
Hua-Yan
Si
*ab,
Du
Yuan
b,
Jing-Sheng
Chen
b and
Gan-Moog
Chow
b
aSchool of Materials Science and Engineering, Shijiazhuang Railway Institute, Shijiazhuang 050043, China. E-mail: dsscnewman@163.com
bDepartment of Materials Science and Engineering National University of Singapore, Kent Ridge, Singapore, 117540, Singapore
First published on 1st September 2011
Abstract
Different sizes of PbS nanocrystals (NCs) were synthesized from sulfur–amine solution. Instead of using a hot-injection method, we prepared alkylamine-PbS NCs at room temperature in air, where the produced PbS NCs were dispersed in a non-polar solvent. In this work, alkylamine played dual roles as both reducing agent and surfactant. The size of PbS NCs could be tuned by controlling the reaction time, the concentration of precursor, the volume ratio of alkylamine to water, as well as the length of alkylamine. This new preparation method to produce PbS NCs especially using shorter length alkylamines such as octylamine as capping molecules, showed great potential in developing nanomaterials for optical devices.
1. Introduction
PbS nanocrystals (NCs) have been the subject of intense research for photonic and optical device applications, such as infrared photovoltaics,1–3 infrared LED,4 FET and IR luminescence.5–7 For these applications, PbS NCs require good dispersibility in organic solvents.8–12 Scientists have been dedicated to finding out simple and practical synthetic routes to generate PbS NCs for many years. Early methods include solution phase,13,14 gas phase,15 solid-state,16polymer films17–19 and glass host fabrication.20–22 Recently it has been reported that the preparation methodvia hot-injection from those for cadmium chalocogenides23–25 permits the good dispersity of PbS NCs in organic solvents. Hexamethyldisilathiane (TMS)6,26,27 is the commonly used source of S in the hot-injection method.6 However, TMS is expensive, toxic and unstable in an open air system. Hyeon's group28,29 and others30–34 replaced TMS with inexpensive sulfur precursors. The oil-phase synthesis also required decomposition of Pb precursors such as oxide, oleate and chloride at high temperatures.6,28,32,35,36 A major common drawback shared by all of the above-mentioned methods is that the experiments were carried out with heating in standard airtight conditions, which increased the complexity and the cost of the synthesis of PbS NCs.In the synthesis of nanocrystals such as Au nanowires, nanorods and nanoparticles,37–39alkylamine serves as reducing agent and decreases the synthesis reaction temperature of nanoparticles.31,40–42 There are a great number of reports working with sulfur–amine solutions.43–48 Although different proposed steps in the “solubilization” of sulfur and liquid amines were found in the literature,31,43,46–48 they all suggested that H2S could be generated effectively. At low temperatures, sulfur-alkylamine solutions presents mainly as alkylammonium polysulfides,48 as shown in reaction 1 in Scheme 1. We further proved this fact by 1H NMR and 13C NMR spectra. Meantime, Davis and Naskshbendi43 had also claimed that the sulfur reacted with the methylene protons α to nitrogen and then formed a thioamide. We propose that maybe there is minor thioamide in the sulfur–alkylamine solutions, in the presence of trace water, the thioamide will be hydrolyzed, releasing hydrogen sulfide as in reactions 2 and 3 in Scheme 1.
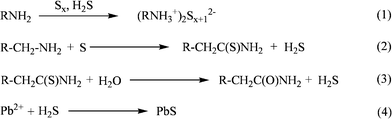 | ||
| Scheme 1 The possible reaction pathways of synthesis of the PbS NCs (R = C6H13, C10H21, C16H32). | ||
To our limited knowledge, there has been no report on using sulfur–alkylamine solution as a sulfur precursor to directly prepare of PbS NCs at room temperature. Herein, we report a room temperature synthesis of monodispersed PbS NCs in air. PbS NCs with different sizes were synthesized in the presence of water and alkylamine (Scheme 1). In our work, alkylamine played dual roles as both reducing agent and surfactant. We observed that nearly cubic PbS NCs could be obtained by using oleylamine at room temperature. This method is different or similar to the previously reported methods in which the PbS NCs were prepared at higher temperatures in either organic or aqueous solvents (hot-injection methods). These include: (i) room temperature synthesis produces large NCs with diameters ranging from 7–17 nm and band-edge absorption features ranging from 1500–2200 nm with full width at half maximum of ∼150 meV. while hot-injection methods produce smaller NCs with diameters ranging from 3–10 nm and band-edge absorption features ranging from 800–1800 nm with FWHM of ∼100 meV;.6,49 (ii) the PbS NCs prepared in our method are truncated cubic while hot-injection methods produce spherical NCs; (iii) the room temperature method produces NCs with a polydispersity similar to or larger than those obtained from hot-injection methods with similar post-synthesis treatments (e.g.precipitation from nonpolar solvents with polar nonsolvents, size-selective precipitation not required). Our method may be used to prepare a large quantity of monodispersed PbS NCs with controlled size at room temperature. The as-prepared NCs were easily dispersed in non-polar organic solution, rendering further treatment easy.
2. Experimental details
In a typical synthesis, elemental sulfur (1 mmol, 32 mg) was added to 10 mL of oleylamine and stirred at room temperature. The color of the solution gradually changed from yellow to orange and formed a homogeneous and clear solution. PbAc2·3H2O (1 mmol, 379 mg) was dissolved in 2.5 mL of water and the resulting lead acetate solution was quickly injected into the sulfur solution at room temperature. The resulting mixture immediately turned black and there appeared sticky material at the bottom of the flask. After the reaction, toluene (5 mL) was added in order to extract the PbS NCs and discarded the sticky materials. The PbS solution was centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 3 min, the precipitate was dissolved in toluene again. The crude product was cleaned by the addition of a small volume of ethanol, centrifugation, and dissolution in toluene. This cleaning procedure was repeated once. The NCs are dispersible in many organic solvents, such as toluene, hexane, and octane.
000 rpm for 3 min, the precipitate was dissolved in toluene again. The crude product was cleaned by the addition of a small volume of ethanol, centrifugation, and dissolution in toluene. This cleaning procedure was repeated once. The NCs are dispersible in many organic solvents, such as toluene, hexane, and octane.
Characterization
X-Ray photoelectron spectroscopy (XPS) was performed on a PHI 5702 spectrometer. X-Ray diffraction (XRD) patterns were measured on a Bruker AXS D8 Advance, Germany. TEM and HRTEM both were carried out on a JEM 3010F, 300 kV. UV-vis absorption spectra were recorded on a Shimadzu UV-3600 UV-vis-NIR spectrophotometer by using pure toluene as reference. The photoluminescence (PL) spectra for IR emission were recorded by using a Horiba Jobin Yyon FluoroLog-3 with an iHR320 attachment equipped with lock-in amplifier and cooled by liquid nitrogen. The InGaAs photodiode detector with detection is limited to 1600 nm. The portion of the PbS NCs PL peak cut off by the detector was fitted using Gaussian. The PL quantum yield (Y) of PbS NCs was determined by referencing to the dye IR125 in DMSO as the standard. The absorbance at the band gap is kept below 0.05 cm−1 to avoid reabsorption of the emitted light. We first determined the linear slope (K) of the plot of fluorescence integrated intensity (F) of the emission peak to the absorbance (A) at the band gap by measuring at least four different concentrations. The PLYPbS is calculated relative to this reference sample, taking the corresponding sample K into account: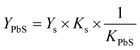
1H NMR and 13C NMR spectra were recorded on a Bruker ACF300 (300 MHz), Bruker DPX300 (300 MHz) spectrometer. Chemical shifts (δ) are reported in ppm relative to TMS (δ 0.00) for the 1H NMR and to chloroform (δ 77.0) for the 13C NMR measurements.
3. Results and discussion
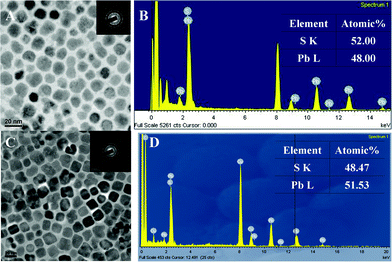
In a typical PbS NCs synthesis, sulfur was dissolved in oleylamine, followed by quick injection of PbAc2 solution. The color of solution mixture changed immediately with observable precipitation. Purified nanocrystals are easily dispersed in nonpolar solvents. A representative TEM image of PbS NCs synthesized using oleylamine is shown in Fig. 1A. The particle shape was truncated cubic. The average particle size was ∼12 nm with a size distribution of 20% (ESI,† Fig. S1). The composition of the PbS samples was identified by energy-dispersive X-ray spectroscopy (EDS) and X-ray photoelectron spectra (XPS) (ESI, Fig. S1 and S2). The HRTEM image of PbS NCs (Fig. 1B) confirmed that each particle was a single crystal. The lattice d-spacing of PbS was estimated to be 0.299 nm, consistent with the (200) lattice planes for bulk PbS (JCPDS Card No. 770244, 2.967 Å). X-Ray powder diffraction pattern also confirmed the crystallinity of PbS nanocrystal (ESI, Fig. S3); the particle size calculated using the Scherrer equation was consistent with TEM results. The absorption spectra for a range of samples are displayed in Fig. 1C, demonstrating tuning from 1500–2200 nm of a well-resolved lowest-energy exciton transition. Compared with the absorption of bulk PbS (3020 nm) a sizable blue-shift occurs. The full width at half maximum of the PbS NC absorption band is ∼150 meV, indicating a low polydispersity of PbS NCs that can be realized without any post-synthesis size-selective precipitation. The spectra featured multiple peaks, which provide further confirmation of the low polydispersity of the NCs. Fig. 1D shows the room-temperature absorption and photoluminescence spectra for a sample containing NCs of about 7 nm in diameter. The intense photoluminescence is primarily due to band-edge emission. (The abrupt drop in intensity for wavelengths higher than 1600 nm is due to the spectral limit of the detector.) Fluorescence quantum yields for these samples were determined to be ∼20% relative to the NIR standard dye IR125 in dimethyl sulfoxide.6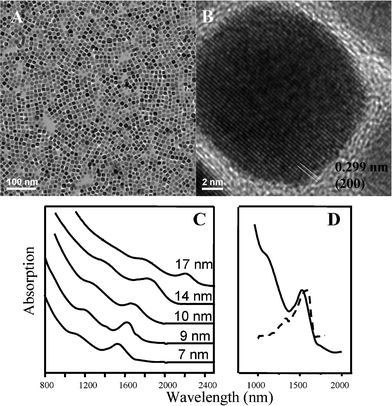 | ||
| Fig. 1 (A) Low-magnification TEM image of PbS NCs; (B) HRTEM image of single PbS NCs. (C) Absorption spectra spanning the range of 7 nm to 17 nm. (D) Band-edge absorption and photoluminescence peaks for a sample ∼7 nm. | ||
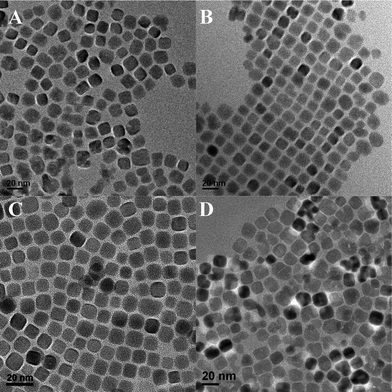
The size of PbS NCs was readily controlled by the reaction time, the concentration, the volume ratio of alkylamine to water and the length of alkylamine. Fig. 2A–C shows the TEM images of PbS NCs with different sizes obtained at reaction time of 15 min, 20 min and 30 min. The gradual increase of size indicated that no additional nucleation occurred during the subsequent growth process (ESI, Fig. S4). As the concentration of PbAc2 increased from 40 mM to 400 mM, the size of PbS NCs decreased from 16 nm to 11 nm, together with a decrease of polydispersity (Fig. 3) (ESI, Fig. S5). When the concentration of PbAc2 increased to 800 mM, the size of PbS NCs exceeded 18 nm and the size and shape became irregular. As the volume ratio of alkylamine to water was changed from 10:1 to 10:2.5, the size of nearly cubic and monodispersed PbS NCs decreased. When the volume ratio of alkylamine to water was further decreased to 10:5, the size of NCs increased to nearly 17 nm (Fig. 4) (ESI, Fig. S6). In the present work, the length of alkylamine was found to be crucial in determining the size and shape of the final products, whereas the size was more influenced than the reaction time, the concentration of the starting material and the volume ratio of alkylamine to water. As shown in Fig. 2D–F, if the concentration, reaction time and volume ratio were kept constant, and oleylamine, dodecylamine and octylamine were used for the reaction, the sizes of the generated NCs were ∼16 nm, ∼13 nm and ∼7 nm, respectively.
 | ||
| Fig. 2 TEM images of (A) 9 nm, (B) 12 nm and (C) 16 nm of oleylamine capped PbS NCs. The size increase was controlled by increasing the reaction time; the effects of the chain length of alkylamine are shown in (D) oleylamine, (E) dodecylamine and (F) octylamine capped PbS NCs. | ||
 | ||
| Fig. 3 TEM images of PbS NCs by changing the concentration of PbAc2: (A) 0.008 mol L−1; (B) 0.04 mol L−1; (C) 0.08 mol L−1 and (D) 0.16 mol/L. | ||
 | ||
| Fig. 4
TEM images of PbS NCs by changing the volume ratio of OA to water: (A) 10:0.5; (B) 10:1; (C) 10:2.5 and (D) 10:5. | ||
An interesting phenomenon was found when as-synthesized PbS NCs was dispersed in a non-polar solvent. Fig. 5 summarizes the shape evolution of PbS NCs in toluene solution. Fig. 5A shows typical TEM images of PbS NCs after reaction, purification, then dispersed in toluene immediately. Only flower-like particles were observed. The enlarged TEM image (inset in Fig. 5A) of a flower showed that the flower was formed by the aggregation of several PbS NCs, each of which is nearly spherical in shape. The aggregation phenomenon may be attributed to the force driven by surface energy. The selected area electron diffraction (SAED) pattern inset in Fig. 5A is consistent with the rock salt structure of bulk PbS materials. After leaving the toluene solution for more than 1 h, the flowers gradually separated, and single particles could be distinguished, as shown in Fig. 5B. At a time of 5 h, aggregated particles detached completely and spherical monodispersed PbS NCs were observed (Fig. 5C). After being in toluene for longer than 24 h, the spherical shaped NCs were found to have become truncated cubic (Fig. 5D). These observations indicate that early growth is kinetically driven, then, with time, thermodynamics gives rise to a truncated cubic shape. This shape transition of PbS NCs was consistent with what other researchers reported earlier.6,50,51 In contrast to previous reports,30,52 we did not find that the primary amine prevented the cube formation as evidenced by TEM images in Fig. 1, in which (200) terminated PbS cubes were formed.
 | ||
| Fig. 5 PbS shapes measured in toluene solution with different setting times: (A) 0 h, (B) 1 h, (C) 5 h, (D) 24 h. | ||
The formation mechanism was investigated. The key to a successful PbS NCs synthesis lies in a controlled nucleation event and subsequent particle growth. When sulfur was dissolved in alkylamine, the color change of the solution indicated sulfur and oleylamine reacted. Fig. S7 (ESI) shows a plot of the 1H NMR and 13C NMR spectra of oleylamine and sulfur–oleylamine. The majority of chemical shifts in sulfur–oleylamine remained the same as those of oleylamine. These results indicated that the large majority of sulfur existed as oleylammonium polysulfides which is consistent with the result of Ozin's group48 (reaction 1 in Scheme 1). The molecular exchange between oleylammonium and oleylamine molecules is expected to be so rapid, due to the very low activation energy of ammonium ion formation, that there are almost no differences between oleylamine and sulfur–oleylamine. Meanwhile, small amounts of sulfur were reduced by alkylamine and formed alkylthioamide (reaction 2 in Scheme 1), which is the same as the formation of octanethioamide reaction proposed by Davis and Naskshbendi.43 However, 1H NMR and 13C NMR spectra are not clear due to little content in our work. The polar group of thioamide in the long alkylamine chain could access water in solution, and would be hydrolyzed, releasing hydrogen sulfide (reaction 3 in Scheme 1). Yoshimura et al.41 and Hodgson et al.45 suggested other ways to produce H2S. Further experiments are to be conducted in order to confirm the step of H2S formation in this reaction. Pb(Ac)2 was chosen as the lead precursor rather than PbCl2, because of its greater solubility and thus improved activity of lead ions. As the Pb(Ac)2 aqueous solution was quickly injected into the sulfur oleylamine solution, the reaction solution color changed quickly, which indicated the PbS NCs nucleated promptly (reaction 4 in Scheme 1), while the alkylamine capping molecules on the PbS NCs coordinated with lead through the –NH2groups.
The IR spectra of the PbS NCs and of oleylamine are similar (Fig. 6), which indicates that oleylamine coordinated the surface Pb atoms with the nitrogen lone pair30 as the capping group. When the oleylamine was replaced by shorter alkylamines such as dodecylamine and octylamine, the size of PbS NCs decreased and the shape varied (Fig. 2D–F), when the other parameters were unchanged. The differences were attributed to the organization of alkylamine and/or dynamics of the monomers in solution, as oleylamine exhibits a “cis” configuration, leading to a shorter extended distance but more prominent steric hindrance than dodecylamine and octylamine. The steric hindrance would prohibit the Pb2+ from accessing the S2− ions, and thus decrease the concentrations of nucleons and increase the sizes of the PbS NCs. Among the three alkylamines used in our work, octylamine had the least steric hindrance effect and increased the activity of the reaction; therefore, the size of PbS NCs was the smallest.
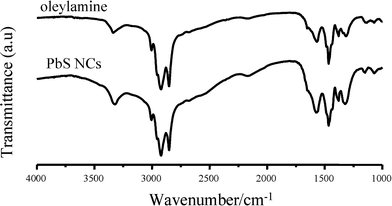 | ||
| Fig. 6 FTIR spectra of oleylamine and PbS NCs. | ||
Below the [Pb2+] concentration of 400 mM and above the alkylamine to water ratio of 10:2.5, the size of PbS NCs decreased as the concentration of [Pb2+] increased and the volume ratio of alkylamine to water decreased (Fig. 3 and 4). Further, the role of acetic acid in adjusting the Pb2+ monomer concentration was investigated. We found that with increased addition of acetic acid to the Pb2+ water solution, the size of PbS NCs increased non-uniformly and the shape turned out to be irregular (Fig. 7). Nucleation took place rapidly immediately after injection and lasted until the monomer concentration dropped below a critical threshold. According to the Ostwald theory, there exists a critical size corresponding to a given monomer concentration. The higher the concentration of the monomer is, the smaller the critical size is. In contrast, the lower the concentration of the monomer is, the larger the critical size is. With increasing the concentration of [Pb2+] or decreasing the volume ratio of alkylamine to water, [Pb2+] increased, so the critical size decreased with increasing the concentration of [Pb2+]. The reactions were terminated before the depletion of monomer concentration, leading to smaller sized and better monodispersed PbS NCs. When acetic acid was added to the lead ionic solution, the concentration of Pb2+ decreased due to re-establishment of the hydrolysis balance. When the monomer concentration was depleted due to growth, the critical size became larger than the average size, and the size distribution broadened viaOstwald ripening. In our experiment, we found the truncated cubic PbS NCs could be obtained without adding acetic acid to the lead ionic solution. When the concentration of [Pb2+] was 800 mM, the [Pb2+] activity was too strong, larger and irregular PbS NCs were produced due to secondary nucleation (Fig. 3). When the volume ratio of oleylamine to water was decreased to 10:5, more water was added into the S–oleylamine solution, and a sticky material was formed, equivalent to reducing the concentration of S2− ions, hence larger PbS NCs were obtained in Fig. 4.
 | ||
| Fig. 7 Adding acetic acid, pH = 4: (A) TEM image and (B) EDS. Adding acetic acid, pH = 3: (C) TEM image and (D) EDS. | ||
Rapid injection of PbS molecular precursor into the reaction solution induced the formation of the tetradecahedron seeds which were terminated by six {100} faces and eight {111} faces50 (Fig. 8), alkylamine preferentially coordinates the {111} faces but is weakly bound to PbS. In the short reaction time, the dynamics determined the shape of PbS NCs due to a large number of monomers, so the shape of freshly prepared PbS NCs was nearly equiaxed when dispersed in non-polar solvent (Fig. 5A and B). However, when increasing the setting time of the PbS NCs solution, the thermodynamics had a prominent effect. The intrinsic surface energy of {111} faces was higher than that of {100} faces, and a relatively fast growth along the <111> directions from the tetradecahedron seeds resulted in the formation of truncated cube-shaped nanocrystals (Fig. 5C and D).
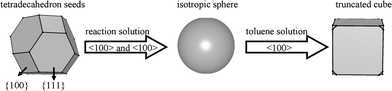 | ||
| Fig. 8 Schematic graph of the structure formation process of PbS NCs. | ||
In the process of synthesis, the alkylamine acted as reducing agent and surfactant. The alkylamine was crucial under this circumstance because it influenced the activity of the monomer species, facilitated the transfer of Pb2+ from the water phase to the oil phase and hence aided in the nanocrystal growth. When oleylamine was replaced by dodecylamine and octylamine (Fig. 2E and F), PbS nanoparticles were also synthesized. Shorter alkylamine as capping ligand on the PbS NCs was more desirable to ease the injection of electrons and holes into the NCs, which improved the electroluminescence efficiency from PbS NCs/polymer composite films.4 In the previously published method, where shorter alkylamine capped PbS NCs were usually attained by the replacement of longer alkyl tails,4,6 the process was tedious and less efficient with unwanted aggregation of NCs. The synthesis method we herein report is more straightforward and efficient, and the final product of monodisperse octylamine capped PbS NCs was obtained.
In conclusion, we have shown a simple and novel method to synthesize PbS nanoparticles. Alkylamine served as reducing agent and surfactant. The size of PbS NCs could be tuned by controlling the reaction time, the concentration of precursor, the volume ratio of alkylamine to water and the length of alkylamine.
Acknowledgements
This work is partially supported by Ministry of Education's AcRF Tier 2 funding T207B1104-RS and Tier 1 funding T11-1001-P04. G.M.C acknowledges the partial support of this work by the US Office of Naval Research. We are grateful to Prof. Wee Shong Chin and Rong Lun Khon in the Department of Chemistry at National University of Singapore for using the photoluminescence in IR measurements.References
- S. A. Mcdonald, G. Konstantatos, S. Zhang, P. W. Cyr, E. J. D. Klem, L. Levina and E. H. Sargent, Nat. Mater., 2005, 4, 138 CrossRef CAS.
- S. Gunesa, K. P. Fritz, H. Neugebauer, N. S. Sariciftci, S. Kumar and G. D. Scholes, Sol. Energy Mater. Sol. Cells, 2007, 91, 420 CrossRef.
- E. J. D. Klem, D. D. MacNeil, P. W. Cyr, L. Levina and E. H. Sargent, Appl. Phys. Lett., 2007, 90, 183113 CrossRef.
- L. Bakuevaa, S. Musikhin, M. A. Hines, T. W. F. Chang, M. Tzolov, G. D. Scholes and E. H. Sargent, Appl. Phys. Lett., 2003, 82, 2895 CrossRef.
- S. Hinds, S. Myrskog, L. Levina, G. Koleilat, J. Yang, S. O. Kelley and E. H. J. Sargent, J. Am. Chem. Soc., 2007, 129, 7218 CrossRef CAS.
- M. A. Hines and G. D. Scholes, Adv. Mater., 2003, 15, 1844 CrossRef CAS.
- L. Bakueva, I. Gorelikov, S. Musikhin, X. S. Zhao, E. H. Sargent and E. Kumacheva, Adv. Mater., 2004, 16, 926 CrossRef CAS.
- E. H. Sargent, Adv. Mater., 2005, 17, 515 CrossRef CAS.
- A. L. Rogach, A. Eychmuller, S. G. Hickey and S. V. Kershaw, Small, 2007, 3, 536 CrossRef CAS.
- A. K. Dutta, T. T. Ho, L. Q. Zhang and P. Stroeve, Chem. Mater., 2000, 12, 1042 CrossRef CAS.
- F. W. Wise, Acc. Chem. Res., 2000, 33, 773 CrossRef CAS.
- Y. Zhou, H. Itoh, T. Uemura, K. Naka and Y. Chujo, Langmuir, 2002, 18, 5287 CrossRef CAS.
- M. T. Nenadovice, M. I. Comor, V. Vasic and O. I. J. Micic, J. Phys. Chem., 1990, 94, 6390 CrossRef.
- T. Trindade, P. O. O'Brien, X. Zhang and M. J. Motevalli, J. Mater. Chem., 1997, 7, 1011 RSC.
- C. Kaito, Y. Saito and K. Fujita, Jpn. J. Appl. Phys., 1987, 26, 1973 CrossRef.
- W. Wang, Y. Liu, Y. Zhan, C. Zheng and G. Wang, Mater. Res. Bull., 2001, 36, 1977 CrossRef CAS.
- S. H. Wang and S. H. Yang, Langmuir, 2000, 16, 389 CrossRef CAS.
- L. S. Li, L. H. Qu, L. J. Wang, R. Lu, X. G. Peng, Y. Y. Zhao and T. J. Li, Langmuir, 1997, 13, 6183 CrossRef CAS.
- Z. Pan, J. Liu, X. G. Peng, T. J. Li, Z. Wu and M. Zhu, Langmuir, 1996, 12, 851 CrossRef CAS.
- N. F. Borrelli and D. W. J. Smith, J. Non-Cryst. Solids, 1994, 180, 25 CrossRef CAS.
- A. Lipovskii, E. Kolobkova, V. Petrikov, I. Kang, A. Olkhovets, T. Krauss, M. Thomas, J. Silcox, F. Wise, Q. Shen and S. Kycia, Appl. Phys. Lett., 1997, 71, 3406 CrossRef CAS.
- A. A. Lipovskii, E. V. Kolobkova, A. Olkhovets, V. D. Petrikov and F. Wise, Phys. E., 2000, 5, 157 CrossRef.
- C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706 CrossRef CAS.
- Z. A. Peng and X. G. Peng, J. Am. Chem. Soc., 2001, 123, 183 CrossRef CAS.
- L. Qu, Z. A. Peng and X. G. Peng, Nano Lett., 2001, 1, 333 CrossRef CAS.
- J. S. Lee, E. V. Shevchenko and D. V. Talapin, J. Am. Chem. Soc., 2008, 130, 9673 CrossRef CAS.
- T. Y. Liu, M. J. Li, J. Y. Ouyang, M. B. Zaman, R. B. Wang, X. Wu, C. S. Yeh, Q. Lin, B. Yang and K. Yu, J. Phys. Chem. C, 2009, 113, 2301 CAS.
- J. Joo, H. B. Na, T. Yu, J. H. Yu, Y. W. Kim, F. X. Wu, J. Z. Zhang and T. Hyeon, J. Am. Chem. Soc., 2003, 125, 11100 CrossRef CAS.
- S. H. Choi, K. AN, E. G. Kim, J. H. Yu, J. H. Kim and T. Hyeon, Adv. Funct. Mater., 2009, 19, 1645 CrossRef CAS.
- L. Cademartiri, J. Bertolotti, R. Sapienza, D. S. Wiersma, G. Freymann and G. A. Ozin, J. Phys. Chem. B, 2006, 110, 671 CrossRef CAS.
- J. C. Liu, H. Z. Yu, Z. L. Wu, W. L.iWang, J. B. Peng and Y. Cao, Nanotechnology, 2008, 19, 345602 CrossRef.
- H. B. Li, D. Chen, L. L. Li, F. Q. Tang, L. Zhang and J. Ren, CrystEngComm, 2010, 12, 1127 RSC.
- J. H. Warner, E. Thomsen, A. R. Watt, N. R. Heckenberg and H. Rubinsztein-Dunlop, Nanotechnology, 2005, 16, 175 CrossRef CAS.
- D. V. Talapin, A. L. Rogach, I. Mekis, S. Haubold, A. Kornowski, M. Haase and H. Weller, Colloids Surf., A, 2002, 202, 145 CrossRef CAS.
- W.-K. Koh, Y. M. Yoon and C. B. Murray, Chem. Mater., 2011, 23, 1825 CrossRef CAS.
- W.-K. Koh, A. C. Bartnik, F. W. Wise and C. B. Murray, J. Am. Chem. Soc., 2010, 132, 3909 CrossRef CAS.
- L. Polavarapu and Q. H. Xu, Nanotechnology, 2009, 20, 185606 CrossRef.
- Z. Li, J. Tao, X. Lu, Y. Zhu and Y. Xia, Nano Lett., 2008, 8, 3052 CrossRef CAS.
- Z. Y. Huo, C. K. Tsung, W. Y. Huang, X. F. Zhang and P. D. Yang, Nano Lett., 2008, 8, 2041 CrossRef CAS.
- L. H. Qu, Z. A. Peng and X. G. Peng, Nano Lett., 2001, 1, 333 CrossRef CAS.
- G. G. Yordanov, H. Yoshimura and C. D. Dushkin, Colloid Polym. Sci., 2008, 286, 813 CAS.
- F. Zhao, M. Rutherford, S. Y. Grisham and X. G. Peng, J. Am. Chem. Soc., 2009, 131, 5350 CrossRef CAS.
- R. E. Davis and H. F. Naskshbendi, J. Am. Chem. Soc., 1962, 84, 2085 CrossRef CAS.
- F. P. Daly and C. W. Brown, J. Phys. Chem., 1973, 77, 1859 CrossRef CAS.
- W. G. Hodgson, S. A. Buckler and G. Peters, J. Am. Chem. Soc., 1963, 85, 543 CrossRef CAS.
- T. G. Levi, Gazz. Chim. Ital., 1930, 60, 975 CAS.
- T. G. Levi, Gazz. Chim. Ital., 1931, 61, 286 CAS.
- J. W. Thomson, K. Nagashima, P. M. Macdonald and G. A. Ozin, J. Am. Chem. Soc., 2011, 133, 5036 CrossRef CAS.
- I. Moreels, Y. Justo, B. D. Geyter, K. Haustraete, J. C. Marins and Z. Hens, ACS Nano, 2011, 5, 2004 CrossRef CAS.
- S. M. Lee, Y. W. Jun, S. N. Cho and J. Cheon, J. Am. Chem. Soc., 2002, 124, 11244 CrossRef CAS.
- S. M. Lee, S. N. Cho and J. Cheon, Adv. Mater., 2003, 15, 441 CrossRef CAS.
- K. S. Cho, D. V. Talapin, W. Gaschler and C. B. Murray, J. Am. Chem. Soc., 2005, 127, 7140 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S7 and Table 1. See DOI: 10.1039/c1ra00279a |
| This journal is © The Royal Society of Chemistry 2011 |