Synthesis of piperitone-derived halogenated lactones and their effect on aphid probing, feeding, and settling behavior†‡
Aleksandra
Grudniewska
a,
Katarzyna
Dancewicz
b,
Agata
Białońska
c,
Zbigniew
Ciunik
c,
Beata
Gabryś
*b and
Czesław
Wawrzeńczyk
*a
aDepartment of Chemistry, Wrocław University of Environmental and Life Sciences, Norwida 25, 50-375 Wrocław, Poland. E-mail: czeslaw.wawrzenczyk@up.wroc.pl; Fax: +48 71 3284124; Tel: +48 71 3205257
bDepartment of Biology and Ecology, University of Zielona Góra, Szafrana 1, 65-516 Zielona Góra, Poland
cFaculty of Chemistry, University of Wrocław, F. Joliot-Curie 14, 50-383 Wrocław, Poland
First published on 17th August 2011
Abstract
Five racemic and five enantiomeric pairs of new halolactones with the p-menthane system were obtained in two or three step synthesis from racemic and optically active cis- and trans-piperitols. The key steps of the syntheses were the Claisen-Johnson rearrangement of piperitols to γ,δ-unsaturated esters and haloloctonization of γ,δ-unsaturated esters or acids. The structures of the compounds were confirmed by spectroscopic and crystallographic data. Antifeedant activity of all iodo-, bromo- and chlorolactones and racemic piperitone against Myzus persicae was examinated. The effect of these compounds on probing, feeding, and settling behavior of M. persicae in vivo was studied.
Introduction
Naturally occurring and synthetic isoprenoid lactones are known for their broad variety of biological activity such as antifungal,1,2 antibacterial,2,3 antitumor4–7 and antifeedant behavior.8–13 Our interest in the synthesis of this group of compounds is connected with their feeding deterrent activity against insects.14–21The most well known antifeedants belong to different chemical groups and come from natural sources,22–24 but the most widespread are those with an isoprenoid structure and lactone moiety, the sesquiterpenoid lactones.8,11 However, the application of natural antifeedants for the protection of plants and stored products against insect pests is limited. The main constraint is the low content in natural sources. Moreover, natural antifeedants usually have complicated structures and their synthesis is complex and also very expensive. Therefore, the synthesis of natural antifeedants' analogues is one of the most promising ways leading to their practical use in insect pest population control.
Following this idea, we have synthesized a number of terpenoid lactones from natural terpenes25,26 and terpenoid alcohols16 and ketones.27–29 Being conscious of the effect of the chiral center configuration of a molecule on the compound activity, we have synthesized, whenever it was possible, all isomers of the final products. Some of the obtained lactones exhibited antifeedant activity comparable with the activity of the most known and active natural antifeedant – azadirachtin.14–16,19,20 Many of the lactones synthesized were also tested for antifeedant activity against aphids.17,18
Aphids (Homoptera, Aphididae) are insects of significant economic importance. The direct damage by aphid feeding is responsible for approximately 2% of total world crop losses caused by insects.30 At the same time, aphids transmit nearly 30% of all known plant virus species and 50% of insect-borne ones.31 Plant diseases caused by these viruses may destroy up to 80% of the yield of a given crop.32 Aphids may acquire and inoculate viruses during various stages of plant penetration with sucking-piercing mouthparts. During brief intracellular probes in epidermis and parenchyma (mesophyll in leaves) that precede feeding in phloem vessels, aphids may transmit non-persistent and semi-persistent viruses. When aphid stylets reach sieve elements, persistent viruses may be transmitted.33–36 It is crucial then, to deter aphid probing or at least prevent feeding, to protect plants from pathogen infection and limit the virus spread within the field crops. Of over 190 aphid species that are known to transmit viruses, the peach-potato aphid Myzus persicae (Sulz.) is of exceptional importance to agriculture.31 It is cosmopolitan, highly polyphagous, and very effective as virus vector. It can infest plants of more that 40 families and transmit more than 100 plant viruses.37
At present, aphid control depends mainly on the use of insecticides. Due to the repeated applications, many aphid species, especially M. persicae, have developed resistance to several aphicides. Therefore, an alternative method of aphid control is needed and the use of targeted chemicals that would repell aphids or deter their probing and feeding is one of the most promising approaches. For example, the sesquiterpenoid polygodial was successfully applied in the field against bird cherry-oat aphid Rhopalosiphum padi L., giving similar results to those obtained with cypermethrin.38
Here we present the stereoselective synthesis of five racemic and five enantiomeric pairs of new halolactones with the p-menthane system. The lactones were obtained from racemic and enantiomerically enriched (ee = 91–98%) cis- and trans-piperitols, which were synthesized from piperitone as described earlier.39 The final compounds were examined for antifeedant activity. The effect of piperitone and piperitone-derived lactones on probing, feeding, and settling behavior of M. persicae was studied in vivo and the physiological and behavioral outcomes were discussed in respect to epidemiological consequences.
Results and discussion
Chemistry
Five racemic and five enantiomerically enriched pairs of new halolactones with the p-menthane system were obtained in two or three step synthesis from racemic and optically active cis- (Scheme 1) and trans-piperitols (Scheme 2). | ||
| Scheme 1 Synthesis of iodolactones 4a–c and bromolactones 5a–c. Reagents: (i) CH3C(OEt)3, CH3COOH, 138 °C; (ii) 1. KOH, MeOH, 2. HCl; (iii) I2, KI, NaHCO3, H2O, Et2O; (iv) Br2, NaHCO3, H2O, Et2O. | ||
 | ||
| Scheme 2 Synthesis of iodolactones 9a–c, bromolactones 10a–c and chlorolactones 11a–c. Reagents: (i) CH3C(OEt)3, CH3COOH, 138 °C; (ii) 1. KOH, MeOH, 2. HCl; (iii) I2, KI, NaHCO3, H2O, Et2O; (iv) Br2, NaHCO3, H2O, Et2O; (v) NCS, THF, H2O. | ||
The first step of the synthesis was the Claisen-Johnson rearrangement40 of corresponding allylic alcohols (1a–c and 6a–c). The structures of the products obtained (2a–c, 7a–c) were determined on the basis of their spectral data. In the 1H NMR spectra of 2a–c a triplet (J = 7.1 Hz, 1.25 ppm) and quartet (J = 7.1 Hz, 4.10 ppm) of ethoxy group and singlet (2.25 ppm) of methylene protons CH2-2 were observed. The multiplets at 5.49–5.54 ppm of H-2′ and H-3′ confirmed the presence of the double bond in the esters 2a–c. In an analogous manner, the structures of esters 7a–c were proved. Unfortunately, spectroscopic data did not allow the determination of the configuration (cis or trans) of the carboethoxymethylene group in relation to the isopropyl group. However, taking into consideration the mechanism of Claisen-Johnson rearrangement of allylic alcohols the relative configuration of substitutents in the molecules of the γ,δ-unsaturated esters obtained can be proposed. According to the mechanism of Claisen-Johnson rearrangement starting from optically active (1a, 1b) and racemic cis-piperitols (1c) the esters (2a, 2b and 2c, respectively) with the cis-oriented carboethoxymethylene group in the relation to the isopropyl group should be obtained (Scheme 3). trans-Piperitols (6a, 6b and 6c) should be transformed into the esters (7a, 7b and 7c, respectively) with trans-orientation of the carboethoxymethylene group with respect to the isopropyl group (Scheme 3). These assumptions were confirmed by the X-ray structures of halolactones (Fig. 1–6), which were obtained in the next steps of the synthesis.
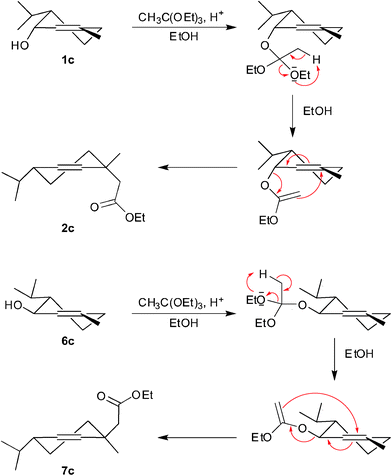 | ||
| Scheme 3 The mechanism of Claisen-Johnson rearrangement of cis- (1c) and trans-piperitol (6c) with triethyl orthoacetate. | ||
Enantiomeric excesses of esters 2a, 2b, 7a and 7b, determined by chiral GC, were the same as the starting alcohols (1a, 1b, 6a and 6b, respectively). These results indicate that the orthoester Claisen rearrangement proceeded in a highly stereospecific manner.40–45 Knowing the absolute configuration of piperitols 1a, 1b, 6a and 6b and taking into consideration the mechanism of Claisen-Johnson rearrangement, it was possible to ascribe the absolute configuration at C-1′ in esters 2a and 7a as S and in esters 2b and 7b as R.
In the next step of the synthesis, the γ,δ-unsaturated esters 2a–c and 7a–c were hydrolyzed to the corresponding acids (3a–c, 8a–c) in methanolic KOH solution. In the IR spectra of the products, the characteristic absorption bands of carboxy group (2600–3500 cm−1 of 3a–c and 8a–c) were observed. The lactonization of cis-γ,δ-unsaturated acids 3a–c with aqueous I2/KI solution in diethyl ether under basic conditions46 afforded corresponding iodolactones (4a–c, respectively). The structure of these products was confirmed by their spectral data. The presence of the γ-lactone ring was confirmed by the absorption band at ν = 1779 cm−1 in the IR spectrum. The value (9.2 Hz) of the coupling constant between H-5 and H-6 protons in the 1H NMR spectrum indicates trans-diaxial orientation of these protons and consequently the equatorial positions of C–I as well as C–O bonds. The presence of the large coupling constant of H-4 with neighboring axial proton H-5 (J = 11.9 Hz) proved its axial orientation and equatorial orientation of the isopropyl group. The X-ray structure of racemic iodolactone 4c (Fig. 1) confirmed the assignation presented above.
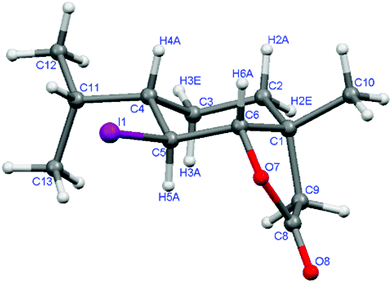 | ||
| Fig. 1 One of two crystallographically unrelated molecules of the crystal structure of iodolactone 4c. | ||
Similarly to the synthesis of racemic lactone 4c, the optically active iodolactones 4a and 4b from corresponding acids (3a and 3b, respectively) were obtained. Their enantiomeric excesses determined by chiral GC were 97% and 96% of isomer 4a and 4b, respectively. The absolute configurations of the products obtained (4a, 4b) were assigned on the basis of structure of substrates and taking into account the stereochemistry of the iodolactonization.46–51 The absolute configurations of 4a and 4b, determined in this way, were fully confirmed by the X-ray crystal structures (Fig. 2).
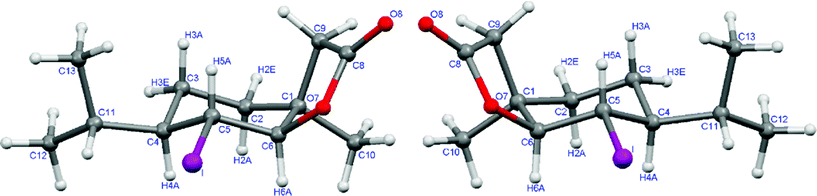 | ||
| Fig. 2 The crystal structures of iodolactones 4a (left) and 4b (right). | ||
In a similar manner to the synthesis of iodolactones 4a–c, the lactonization of trans-γ,δ-unsaturated acids 8a–c afforded corresponding iodolactones (9a–c, respectively). Their structures were also confirmed by NMR and IR spectroscopic data. The absorption band characteristic for the γ-lactone ring is present in the IR spectra of 9a–c at 1775 cm−1. In the 1H NMR spectra of 9a–c two multiplets of H-5 and H-6 at 4.63 and 4.69 ppm were present. The small value of the coupling constants in these signals proved the trans-diequatorial orientation of coupled protons. Therefore, it can be concluded that C–I as well as C–O bonds are located in the axial positions. The HSQC (heteronuclear single quantum coherance) experiment showed that the signal at 4.69 correlates with the signal at 87.88 ppm of carbon atom directly connected to the oxygen atom. On the basis of these data the multiplets at 4.69 and 4.63 ppm were assigned to the protons H-6 and H-5, respectively. The full confirmation of the structure of 9c was provided by the X-ray crystal structure as shown in Fig. 3. The absolute configuration of enantiomerically enriched iodolactones 9a (ee = 98%) and 9b (ee = 94%), similarly to the lactones 4a and 4b, were assigned on the basis of the structure of the substrates used and taking into account the stereochemistry of the iodolactonization.
 | ||
| Fig. 3 The crystal structure of iodolactone 9c. | ||
As a result of lactonization of γ,δ-unsaturated carboxylic acids 3a–c and 8a–c, using bromine as an electrophilic agent, the racemic and optically active bromolactones 5a–c and 10a–c were obtained, respectively (Scheme 1 and 2). The structures of the products obtained were determined on the basis of spectral data. In the IR spectra of lactones 5a–c and 10a–c, the characteristic absorption bands of the γ-lactone ring (1779 and 1780 cm−1, respectively) were observed. The presence of doublet (δ = 4.33) of H-6 with coupling constant (J = 8.7 Hz) with neighboring proton H-5 in the 1H NMR spectra of 5a–c proved their trans-diaxial orientation and consequently the equatorial positions of C-Br as well as C-O bonds. Additionally, the values (JH4-H5 = 11.7 Hz, JH6-H5 = 8.7 Hz) of two coupling constants found in doublet of doublets at 3.69 ppm of H-5 proton indicates the axial orientation of H-4 and therefore the trans-diequatorial positions of the bromine atom and the isopropyl group. The enantiomeric excesses of bromolactones 5a and 5b determined by chiral GC were 97% and 91%, respectively. Since the lactones 5a and 5b were nice crystals, the performing of the X-ray crystal structure analysis was possible. This analysis confirmed the absolute configurations of chiral centers which were assigned on the basis of the structure of substrates and taking into account the stereochemistry of the bromolactonization as (1S,4R,5R,6R) for the lactones 5a and as (1R,4S,5S,6S) for 5b (Fig. 4).
 | ||
| Fig. 4 The crystal structures of bromolactones 5a (left) and 5b (right). | ||
The presence of two signals at 4.60 and 4.54 ppm in the 1H NMR spectra of δ-bromo-γ-lactones obtained from trans-piperitols (10a–c) proved that both γ-lactone ring and C-Br bond were formed. The shapes of these signals suggests, that the multiplet like triplet at 4.60 ppm is from proton H-5 and doublet (J = 2.3 Hz) at 4.54 ppm is from H-6. These assignments were confirmed by HSQC experiment, which showed that the signal at 4.54 ppm correlates with the signal at 86.26 ppm of the carbon atom directly connected to the oxygen atom. Furthermore, the small values of the coupling constants in the multiplets of H-5 and H-6 proved trans-diequatorial orientation of these protons. All these data were clearly confirmed by the crystal structure obtained for 10c (Fig. 5). The enantiomeric excesses (according to chiral GC) of bromolactones 10a and 10b, obtained from corresponding acids (8a and 8b) (Scheme 2), were 94% and 96%, respectively.
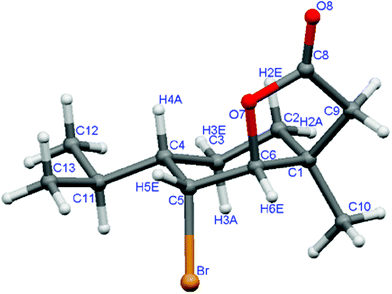 | ||
| Fig. 5 The crystal structure of bromolactone 10c. | ||
To obtain chlorolactones, the reaction of γ,δ-unsaturated esters 2a–c and 7a–c with N-chlorosuccinimide (NCS) was carried out. In the reaction of cisesters 2a–c with NCS, a mixture (according to GC) of many products was formed. Unfortunately, it was impossible to separate them and consequently determine their structures. A similar situation was observed in the trials of the lactonization of these esters with NBS and NIS. However, in the reaction of trans-γ,δ-unsaturated esters 7a–c with NCS, the corresponding chlorolactones 11a–c were obtained as the only products. The structures of these lactones were determined on the basis of spectral data. The IR spectra of 11a–c (1785 cm−1) confirmed that the γ-lactone ring was formed. The value (J = 2.3 Hz) of the coupling constant between protons H-5 (4.54 ppm) and H-6 (4.38 ppm) found in the 1H NMR spectra indicates that they are in trans-diequatorial position. Our assignments were fully confirmed by X-ray crystallography (Fig. 6). Lactonization stereospecificity confirms that the enantiomeric excesses of lactones 11a (ee = 91%) and 11b (ee = 96%), determined by chiral GC, were identical to the enantiomeric excesses of esters (7a and 7b respectively) which were used for the reaction.
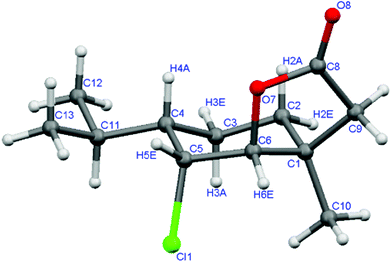 | ||
| Fig. 6 One of two crystallographically unrelated molecules of the crystal structure of chlorolactone 11c. | ||
Biology
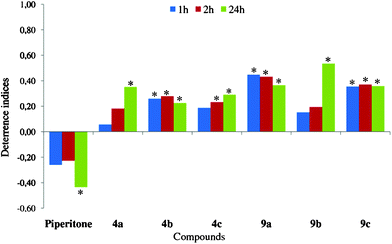
The feeding process of insects can be restrained at three levels: preingestional (immediate effect associated with host finding and host selection processes involving gustatory receptors), ingestional (related to food transport and production, release, and digestion by salivary enzymes), and postingestional (long-term effects involving various aspects of digestion and absorption of food).52 In aphids, the alteration of behaviour during probing in epidermis and mesophyll before the first phloem phase points at the effect of a preingestional factor. The high proportion of short (<10 min) probes among all probes, the long periods of non-penetration, and a low success rate in finding sieve elements are the most important symptoms of preingestional deterrence. Similarly, any disturbances in sap uptake from sieve elements, such as the reduction of ingestion periods and long periods of salivation during the phloem phase can be attributed to ingestional effect of a sieve element-associated deterrent. The refusal to settle on plants even if the feeding process is not impeded may be a sign of postingestional deterrence.17 Beside the direct negative effect on aphid feeding, a deterrent that impedes preingestional and ingestional activities should also prevent the transmission of non-persistent and persistent viruses, respectively.In the present study, the natural monoterpene ketone – piperitone appeared to be an attractant to the peach potato aphid Myzus persicae. The duration of probing and proportion of phloem phase in total probing were similar in aphids on control and piperitone-treated leaves. All aphids reached phloem vessels and almost all of them showed sustained phloem sap ingestion during the first probe. The salivation – ingestion ratio during probing in sieve elements was five times lower than in aphids on control leaves. The prolonged phloem salivation would have indicated the aphid response to negative chemical factors in the plant sap.17 24 h after application, aphids were clearly attracted to settle on piperitone-treated leaves (Fig. 7–10; Tables 1 and 2).
 | ||
| Fig. 7 Effect of iodolactones (4a–c and 9a–c) on settling of Myzus persicae on plants. Deterrence indices after 1, 2, and 24 h. *Significant differences in aphid settling on lactone-treated vs. control leaves (P < 0.05). | ||
 | ||
| Fig. 8 Effect of bromolactones (5a–c and 10a–c) on settling of Myzus persicae on plants. Deterrence indices after 1, 2, and 24 h. *Significant differences in aphid settling on lactone-treated vs. control leaves (P < 0.05). | ||
 | ||
| Fig. 9 Effect of chlorolactones (11a–c) on settling of Myzus persicae on plants. Deterrence indices after 1, 2, and 24 h. *Significant differences in aphid settling on lactone-treated vs. control leaves (P < 0.05). | ||
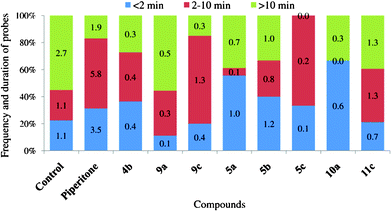 | ||
| Fig. 10 Frequency and duration of probes before the first phloem phase after application of piperitone and halolactones. Numbers represent number of probes that fall into three categories: shorter than 2 min, 2–10 min long, and longer than 10 min. | ||
| Non-probing [h] | Pathway phase [h] | Phloem phase [h] | Number of probes | Number of probes with phloem phase | |
|---|---|---|---|---|---|
| a A and B denote statistically significant differences in comparison to control and piperitone, respectively (ANOVA, P < 0.05). | |||||
| Control | 0.4 (±0.1) | 4.0 (±0.7) | 3.6 (±0.8) | 6.8 (±1.6) | 1.3 (±0.3) |
| Piperitone | 0.6 (±0.1) Aa | 4.2 (±0.5) | 3.2 (±0.5) | 19.1 (±3.9) A | 1.7 (±0.2) |
| Iodolactones | |||||
| 4b | 0.6 (±0.2) | 3.5 (±0.6) | 3.9 (±0.7) | 7.3 (±1.6) | 3.7 (±0.4) AB |
| 9a | 0.5 (±0.2) | 2.7 (±0.4) B | 4.7 (±0.5) B | 4.5 (±0.9) B | 1.9 (±0.3) |
| 9c | 3.0 (±0.7) AB | 2.6 (±0.4) B | 2.4 (±0.6) | 10.7 (±1.8) | 3.7 (±0.8) AB |
| Bromolactones | |||||
| 5a | 0.5 (±0.3) B | 5.7 (±0.5) B | 1.8 (±0.5) B | 7.2 (±3.7) B | 3.0 (±1.7) |
| 5b | 1.4 (±0.7) | 5.0 (±0.9) | 1.6 (±0.7) AB | 9.0 (±1.8) B | 1.2 (±0.3) |
| 5c | 1.2 (±0.7) | 6.0 (±0.7) B | 0.8 (±0.5) AB | 2.8 (±1.0) AB | 1.1 (±0.2) |
| 10a | 1.6 (±0.9) | 5.6 (±0.9) | 0.8 (±0.5) AB | 3.8 (±1.7) AB | 0.7 (±0.2) B |
| Chlorolactones | |||||
| 11a | 1.3 (±0.4) A | 5.0 (±0.5) | 1.7 (±0.5) B | 14.1 (±3.2) | 1.3 (±0.3) |
| Aphids with Ea[%] | Aphids with E>10 in 1st probe [%] | E2>10 minb [#] | E/(C+E)cd [%] | E1/(E1+E2)de [%] | |
|---|---|---|---|---|---|
| a Aphids that reached phloem vessels during the 8-hour experiment. b Periods of sustained phloem sap ingestion. c Proportion of phloem phase during stylet penetration. d Aphids that did not reach phloem elements during 8-hour experiment were excluded from this analysis. e Proportion of phloem salivation to total phloem phase. f A and B denote statistically significant differences in comparison to control and piperitone, respectively (ANOVA, P < 0.05). | |||||
| Control | 92 | 50 | 1.8 (±0.3) | 50.5 (±10,0) | 12,1 (8.9) |
| Piperitone | 100 | 92 | 2.2 (±0.3) | 42,7 (±6.7) | 2.5 (±1.1) |
| Iodolactones | |||||
| 4b | 100 | 27 | 2.6 (±0.6) | 53.5 (9.8) | 25.9 (6.8)AB |
| 9a | 100 | 33 | 2.5 (±0.3) | 62.4 (5.8) Bf | 12.9 (4.0) B |
| 9c | 92 | 17 | 2.5 (±0.6) | 46.1 (8.1) | 27.4 (8.4) AB |
| Bromolactones | |||||
| 5a | 92 | 17 | 0.9 (±0.3) | 25.0 (6.6) A | 32.5 (±12.0) AB |
| 5b | 75 | 33 | 0.5 (±0.2) | 26.8 (11.9) | 45.0 (±13.2)B |
| 5c | 83 | 25 | 0.3 (±0.1) | 12.9 (6.7) AB | 46.5 (±13.2) AB |
| 10a | 55 | 0 | 0.2 (±0.1) | 15.4 (9.7) AB | 57.4 (19.4) AB |
| Chlorolactones | |||||
| 11a | 75 | 58 | 1.3 (±0.4) | 31.9 (±7.7) | 6.8 (±3.2) |
The introduction of a lactone moiety and halogen atom into a piperitone molecule in the halogenation process dramatically changed its biological activity (Fig. 7–9). The chlorolactones (11a–c) had the strongest deterrent effect on aphid settling (DI24: 0.78–0.93), the bromolactones (5a–c and 10a–c) were very strong (DI24: 0.45–0.84), and the iodinated ones were the weakest ones (DI24: 0.23–0.53) (Fig. 9, 8 and 7, respectively). All chloro- and bromolactones that originated from pure (‘+’ or ‘−’) enantiomers of either cis- or trans-piperitol had the strongest and the most durable deterrent effect. They deterred aphid settling from the onset of experiment up to at least 24 h (end of experiment) and their deterrence indices were relatively high. Chloro- (11c) and bromolactones (10c) that were obtained from racemic trans-piperitol showed a delayed deterrent effect. Compounds with chiral centres configurations: 1S,4S,5R,6R (chlorolactone 11a), 1S,4R,5R,6R (bromolactone 5a), and 1R,4S,5S,6S (bromolactone 5b) were the strongest deterrents in aphid settling bioassay. The indices of deterrence of iodinated compounds did not differ considerably among members of this group. Nevertheless, the iodolactones derived from trans-piperitol (9a–c) were slightly stronger deterrents than those obtained from the cis-isomer (4a–c) (Fig. 7).
The aphid probing behaviour was monitored using the EPG (Electrical Penetration Graph) technique after the application of piperitone and the following halogenated lactones: chlorolactone 11a, bromolactones 5a–c and 10a, and iodolactones 4b, 9a and 9c. These lactones combined three values discovered in the aphid settling bioassay: quickness of action (deterrent activity expressed as soon as 1 h after application), durability (deterrent activity expressed for as long as 24 h), and potency (relatively high value of DI24 – deterrence index 24 h after application). The EPG recording revealed all kinds of aphid activities related to plant penetration: non-probing, pathway phase (‘C’), derailed stylet movements ‘F’, phloem salivation and sap ingestion ‘E1’ and ‘E2’, respectively, and xylem sap uptake ‘G’. ‘F’ and ‘G’ activities occurred sporadically irrespective of a treatment. Therefore, in statistical analysis, these activities were included in the pathway phase ‘C’ that represents stylet penetration in non-phloem plant tissues.
The aphid total probing time was significantly shorter on chlorolactone 11a- and iodolactone 9c-treated leaves than on control ones (Table 1). The number of probes was significantly lower on bromolactone-treated leaves. Among the probes before the first phloem phase on bromolactone-treated leaves, the short, 2 and 2–10 min probes, predominated. The very low number of probes longer than 10 min on 5c-treated leaves strongly suggests that deterrent factors located in epidermis and mesophyll caused the discontinuation of probing. In this experiment, aphids were tethered, which means that if they had been free to move, they would have probably walked away from the unsuitable feeding site. On control and 9a-treated leaves, the probes longer than 10 min comprised more than 50% of all probes (Fig. 10). The total duration of pathway, i.e. stylet activities in peripheral plant tissues was similar in all aphids irrespective of a treatment. However, in relation to aphids on piperitone-treated leaves, there was a tendency to shorten pathway time after application of iodolactones 9a and 9c and lengthen after application of bromolactones 5a and 5c.
The total duration of phloem phase was the longest in aphids on control, piperitone, and 9c-treated leaves (50, 62, and 43% of the probing time, respectively) (Table 2). The duration of phloem activities was significantly reduced in aphids on bromolactone-treated leaves (down to 13–27% of total probing) in relation to aphids on control as well as piperitone-treated leaves (Table 2). On bromolactone-treated leaves (esp. 10a-treated) there was the highest proportion of aphids that did not succeed in reaching phloem vessels during the 8-hour experiment and the lowest proportion of aphids that showed phloem activities during the 1st probe. In contrast, the number of periods of sustained sap ingestion was similar in all aphids. The proportion of salivation during sieve element-related aphid activities was the lowest on piperitone-treated leaves (2.5%) and the highest on bromolactone- and iodolactone-treated leaves (33–57 and 26–27%, respectively) (Tables 1 and 2).
In summary, the chlorolactones were in all probability not deterrent in a gustatory manner but had a postingestional and delayed effect on aphid behaviour. The total duration of probing and phloem phase was not reduced, the individual probes were not impeded, the salivation into sieve elements was limited, and almost all aphids that reached phloem vessels ingested sap in a sustained manner (i.e., for periods longer than 10 min) during the first probe. Nevertheless, all chlorolactones significantly prevented the aphids from settling on the treated leaves and the potency of the deterrent effect increased in the course of the 24 h experiment.
The bromolactones had most likely preingestional and ingestional deterrent effect on aphids. The phloem phase was 2–10 times shorter than in aphids on control as well as piperitone-treated leaves although total duration of probing was not reduced. Nevertheless, the probes made by aphids before the first phloem phase were relatively short (most of them not longer than 10 min), which means that they did not reach beyond mesophyll. The effect was the strongest in aphids on 5c-treated leaves. Many aphids did not reach phloem vessels during the 8-hour experiments, and very few aphids started feeding during the first probe. Phloem salivation made up 33–57% of all phloem activities. An increase in proportion of salivation during phloem phase was noteworthy: up to 5 times higher than in control and up to 23 times higher than on piperitone-trated leaves. At the same time, all bromolactones had a strong and durable settling deterrent effect on aphids. Moreover, its potency increased in the course of time.
Iodolactones were probably ingestional deterrents. Although the total duration of phloem phase was not reduced, the proportion of phloem salivation was relatively high as compared to aphids on control and piperitone-treated leaves. Aphids were deterred from settling on iodolactone-treated leaves.
Conclusion
In summary, a series of novel halolactones with the p-menthane system has been synthesized in order to investigate the antifeedant activity. The high stereoselectivity method of the Claisen rearrangement and halolactonization processes has been proved.All of the iodo-, bromo-, and chlorolactones had a negative effect on probing, feeding, and settling behavior of Myzus persicae, which was in contrast to the natural compound, starting piperitone that showed weak attractant properties. However, the deterrent activity of individual compounds varied in potency, the time of expression, and the duration of the effect, depending, firstly, on the halogen atom that was built into the molecule and, secondly, on spatial structure of the lactone. In respect to epidemiological importance of aphids as virus vectors, the bromolactones, especially 5a and 10a, would be the most promising preingestional and ingestional deterrents to prevent non-persistent, semi-persistent and persistent plant virus transmission.
Experimental section
General methods
Bromine and N-chlorosuccinimide were purchased from Aldrich, triethyl orthoacetate from Fluka, and iodine from POCH S.A. Racemic and optically active cis- and trans-piperitols were synthesized as described earlier.39 Enantiomerically enriched alcohols were obtained from racemic piperitols by their enzymatic (lipase Amano PS) esterification with vinyl propionate in solvent-free system. The enzymatic resolution of racemic cis- and trans-piperitols was performed several times. Enantiomeric excesses of optically active alcohols were as follows: 97–98% for 1a, 91–96% for 1b, 91–98% for 6a and 94–96% for 6b. Analytical TLC were performed on Fluka Kieselgel 60, F254 plates with mixture of hexane, and diethyl ether in various ratios. Compounds were detected by spraying the plates with 1% Ce(SO4)2, 2% H3[P(Mo3O10)4] in 10% H2SO4, followed by heating to 120–200 °C. Column chromatography was performed on silica gel (Kieselgel 60, 230–400 mesh ASTM, Merck) with a mixture of hexane, methylene chloride and diethyl ether (in various ratio) as eluents. Gas chromatography analysis was carried out on an Agilent 6890N GC instrument (FID, carrier gas H2) using the capillary columns Thermo TRACETM TR-5 (30 m × 0.32 mm × 1.0 μm). Chiral gas chromatography was carried out on the following columns: CP-Chirasil-DEX CB (25 m × 0.25 mm × 0.25 μm) for 2a–c, 4a–c, 7a–c, 9a–c, 10a–c, 11a–c and GAMMA DEXTM 225 (30 m × 0.25 mm × 0.25 μm) for 5a–c. Enantiomeric excesses were determined with the following temperature programs: for 2a–c and 7a–c: 90 °C, 130 °C (1 °C min−1), 200 °C (20 °C min−1) (3 min), the total run time was 46.50 min, tR2a = 30.71 min, tR2b = 31.10 min, tR7b = 32.92 min, tR7a = 33.49 min; for 4a–c, 9a–c and 10a–c: 100 °C (1 min), 190 °C (1.6 °C min−1) (1 min), 200 °C (50 °C min−1) (2 min), the total run time was 60.45 min, tR10a = 36.49 min, tR10b = 38.85 min, tR9a = 40.27 min, tR9b = 40.58 min, tR4b = 42.84 min, tR4a = 43.24 min; for 5a–c: 140 °C, 180 °C (2 °C min−1) (25 min), 200 °C (2 °C min−1) (10 min), the total run time was 65.00 min, tR5b = 46.49 min, tR5a = 47.40 min; for 11a–c: 100 °C, 160 °C (1 °C min−1), 200 °C (50 °C min−1) (2 min), the total run time was 62.80 min, tR11a = 42.22 min, tR11b = 42.74 min. 1H NMR, 13C NMR, DEPT 135 (distortionless enhancement by polarisation transfer), 1H–1H COSY (correlation spectroscopy) and HSQC (heteronuclear single quantum correlation) spectra were recorded for CDCl3 solution on a Bruker Avance II 600 MHz spectrometer. Chemical shifts were referenced to the residual solvent signal (δH = 7.26, δC = 77.0). IR spectra were recorded for liquid films or plate in KBr on a Thermo-Nicolet IR300 FT-IR spectrometer. Melting points (uncorrected) were determined on a Boetius apparatus. Optical rotations were determined on an Autopol IV automatic polarimeter (Rudolph) in chloroform solutions, concentrations denoted in g 100 mL−1. CH elemental analyses were performed on a CE Instruments EA-1110 elemental analyzer. X-ray data were collected on a Kuma KM4CCD diffractometer (Mo-Kα radiation; λ = 0.71073 Å) at 100 K using an Oxford Cryosystem device. Data reduction and analysis was carried out with the CrysAlice ‘RED’ program.53 The space group was determined using the XPREP program. Structures were solved by direct methods using the XS program and refined using all F2 data, as implemented by the XL program.54Non-hydrogen atoms were refined with anisotropic displacement parameters. All H atoms were found in Δρ maps or placed at calculated positions. Before the last cycle of refinement all H atoms were fixed and were allowed to ride on their parent atoms. The Friedel pairs were merged before the final refinement.Chemistry
The mixture of allylic alcohol (10 mmol), triethyl orthoacetate (70 mmol) and a catalytic amount of propionic acid (one drop) was heated at 138 °C for 3 h under the conditions for distillative removal of ethyl alcohol. When the reaction was completed (GC, TLC), the excess of triethyl orthoacetate was distilled off and the crude product was purified by column chromatography (hexane–methylene chloride–diethyl ether, 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2). The yields of the reactions, physical and spectral data of the esters obtained are given below:
:2:2). The yields of the reactions, physical and spectral data of the esters obtained are given below:
γ,δ-Unsaturated ester (5 mmol) was dissolved in 15 mL of 2.5% KOH/MeOH solution and 1 mL of water and the mixture refluxed for 2 h. After evaporating the solvent, the residue was diluted with water and organic impurities were extracted with diethyl ether. The water layer was acidified with 0.5 M HCl solution (pH ≈ 4) and extracted with diethyl ether. The ethereal extract was washed with brine, dried over anhydrous MgSO4 and concentrated in vacuo. The crude product was purified by column chromatography (hexane – diethyl ether, 2:1). The yields of the reactions, physical and spectral data of the acids obtained are given below:
A mixture of γ,δ-unsaturated acid (3 mmol) in 20 mL of diethyl ether and 20 mL of 0.5 M NaHCO3 solution was stirred at room temperature for 20 min and then refluxed. To the refluxing mixture, a solution of I2 (6 mmol) and KI (18 mmol) in 12 mL water was added dropwise. The mixture was stirred under reflux for 3 h. After cooling, it was diluted with diethyl ether and washed with saturated Na2S2O3 solution. The separated ethereal solution was washed with 0.5 M NaHCO3 solution, brine, dried over anhydrous MgSO4 and concentrated in vacuo. The crude product was purified by column chromatography (hexane–diethyl ether–methylene chloride 6:1:1). The yields of the reactions, physical and spectral data of the iodolactones obtained are given below:
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 9.690(2), b = 11.404(2), c = 12.212(3) Å, α = 79.09(4), β = 84.20(3), γ = 86.93(3)°, V = 1317.5(5) Å3, Z = 4, Dc = 1.624 Mg m-3, μ = 2.412 mm−1T = 100(2)K, R = 0.022, wR2 = 0.051 (3931 reflections with I > 2σ(I)) for 271 variables; Elemental analysis calcd (%) for C12H19IO2 (322.17): C 44.74, H 5.94; found: C 45.17, H 6.29.
, a = 9.690(2), b = 11.404(2), c = 12.212(3) Å, α = 79.09(4), β = 84.20(3), γ = 86.93(3)°, V = 1317.5(5) Å3, Z = 4, Dc = 1.624 Mg m-3, μ = 2.412 mm−1T = 100(2)K, R = 0.022, wR2 = 0.051 (3931 reflections with I > 2σ(I)) for 271 variables; Elemental analysis calcd (%) for C12H19IO2 (322.17): C 44.74, H 5.94; found: C 45.17, H 6.29.
A mixture of γ,δ-unsaturated acid (3 mmol) in 20 mL of diethyl ether and 20 mL of 0.5 M NaHCO3 solution was stirred at room temperature for 20 min and then cooled in an ice bath. To the ice-cooling and stirring mixture, Br2 (6 mmol) was added dropwise. The reaction temperature was gradually increased to room temperature and the mixture was stirred for 2 h. After this time the reaction mixture was diluted with diethyl ether and washed with saturated Na2S2O3 solution. The separated ethereal solution was washed with 0.5 M NaHCO3 solution, brine, dried over anhydrous MgSO4 and concentrated in vacuo. The crude product was purified by column chromatography (hexane–diethyl ether–methylene chloride 8:1:1). The yields of the reactions, physical and spectral data of the bromodolactones obtained are given below:
To the solution of γ,δ-unsaturated ester (5 mmol) in a mixture of THF (15 mL) and water (6 mL), N-chlorosuccinimide (7 mmol) was added. The mixture was stirred at room temperature for 24 h. Then the reaction mixture was diluted with diethyl ether and washed with 0.5 M NaHCO3 solution. The separated ethereal solution was washed with brine, dried over anhydrous MgSO4 and concentrated in vacuo. The crude product was purified by column chromatography (hexane–diethyl ether 9:1). The yields of the reactions, physical and spectral data of the chlorolactones obtained are given below:
Bioassays
The data were analyzed using one way ANOVA (STATISTICA 6.1. package). If aphids showed clear preference for the leaf treated with the tested compound (P < 0.05), the compound was described as having attractant properties. If aphids settled mainly on the control half of the leaf (P < 0.05), the compound tested in the respective choice-test was stated a deterrent. From the data thus obtained the relative index of deterrence (DI) was calculated:
| DI = (C − T)/(C + T) | (1) |
C – the number of aphids settled on control half of the leaf
T – the number of aphids settled on the treated with tested compound half of the leaf
The value of DI ranged between 1 (ideal deterrent) and –1 (ideal attractant).
One-way analysis of variance (ANOVA) was carried out on the experimental results. All calculations were performed using STATISTICA 6.1 package (StatSoft, Tulsa, OK, USA).
Acknowledgements
This work is a part of research supported by Polish Ministry of Science and Higher Education (Grant No. N N310 146835).References
- C. Koukoulitsa, G. D. Geromichalos and H. Skaltsa, J. Comput.-Aided Mol. Des., 2005, 19, 617–623 CrossRef CAS.
- M. F. Neerman, Int. J. Aromather., 2003, 13, 114–120 CrossRef.
- N. H. Fischer, T. Lau, C. L. Cantrell, J. Castaneda-Acosta, L. Quijano and S. G. Franzblaus, Phytochemistry, 1998, 49, 559–564 CrossRef CAS.
- K. H. Lee, T. Ikuba, D. Sims, O. Muraoka, H. Kiyokawa, I. H. Hall and H. L. Kim, J. Med. Chem., 1981, 24, 924–927 CrossRef CAS.
- V. Dirsch, H. Stuppner and A. M. Vollmar, Planta Med., 2001, 67, 557–559 CrossRef CAS.
- E. Koukoulista, H. Skaltsa, A. Karioti, C. Demetzos and K. Dimas, Planta Med., 2002, 68, 649–652 CrossRef.
- J. Wen, K. R. You, S. Y. Lee, C. H. Song and D. G. Kim, J. Biol. Chem., 2002, 227, 38954–38964 CrossRef.
- J. Cis, G. Nowak and W. Kisiel, Biochem. Syst. Ecol., 2006, 34, 862–867 CrossRef CAS.
- J. Harmata and J. Nawrot, Biochem. Syst. Ecol., 1984, 12, 95–98 CrossRef.
- H. M. R. Hoffman and J. Rabe, Angew. Chem., Int. Ed. Engl., 1985, 24, 94–110 CrossRef.
- J. Nawrot, B. Drożdż and M. Holub, Herba Pol., 1985, 31, 209–212 Search PubMed.
- B. Rodriquez, B. Rodriques, M. C. Torre, M. S. J. Simmonds and W. M. Blaney, J. Nat. Prod., 1999, 62, 594–600 CrossRef.
- M. E. Sosa, C. E. Tonn and O. S. Giordano, J. Nat. Prod., 1994, 57, 1262–1265 CrossRef CAS.
- M. Szczepanik, I. Dams and C. Wawrzeńczyk, Entomol. Exp. Appl., 2008, 128, 337–345 CrossRef CAS.
- M. Szczepanik, I. Dams and C. Wawrzeńczyk, Environ. Entomol., 2005, 34, 1433–1440 CrossRef CAS.
- E. Paruch, Z. Ciunik, J. Nawrot and C. Wawrzeńczyk, J. Agric. Food Chem., 2000, 48, 4973–4977 CrossRef CAS.
- K. Dancewicz, B. Gabryś, I. Dams and C. Wawrzeńczyk, J. Chem. Ecol., 2008, 34, 530–538 CrossRef CAS.
- A. Halarwwicz-Pacan, B. Gabryś, K. Dancewicz and C. Wawrzeńczyk, J. Plant Prot. Res., 2003, 43, 133–142 Search PubMed.
- J. Nawrot, I. Dams and C. Wawrzeńczyk, J. Stored Prod. Res., 2009, 45, 221–225 CrossRef CAS.
- E. Paruch, J. Nawrot and C. Wawrzeńczyk, Pest Manage. Sci., 2001, 57, 776–780 CrossRef CAS.
- C. Wawrzeńczyk, I. Dams, A. Szumny, M. Szczepanik, J. Nawrot, A. Prądzyńska, B. Gabryś, E. Magnucka, B. Gawdzik, R. Obara and A. Wzorek, Pol. J. Environ. Stud., 2005, 14, 69–84 Search PubMed.
- M. Wawrzyniak, Pol. Pismo Entomol., 1996, 65, 93–96 Search PubMed.
- E. A. Klein Gebbinck, B. J. M. Jansen and A. de Groot, Phytochemistry, 2002, 61, 737–770 CrossRef CAS.
- C. Wawrzeńczyk, E. Paruch and T. Olejniczak, in Chemical Products in Agriculture and Environment, ed. H. Górecki and Z. Dobrzański, Czech-Pol Trade, Prague, 2002, pp. 206–213 Search PubMed.
- E. Paruch, Z. Ciunik and C. Wawrzeńczyk, Eur. J. Org. Chem., 1998, 2677–2682 CrossRef CAS.
- S. Lochyński, B. Frąckowiak, T. Olejniczak, Z. Ciunik and C. Wawrzeńczyk, Tetrahedron: Asymmetry, 2002, 13, 1761–1767 CrossRef.
- I. Dams, A. Białońska, Z. Ciunik and C. Wawrzeńczyk, J. Agric. Food Chem., 2004, 52, 1630–1634 CrossRef CAS.
- I. Dams, A. Białońska, Z. Ciunik and C. Wawrzeńczyk, Eur. J. Org. Chem., 2004, 2662–2668 CrossRef CAS.
- I. Dams, A. Białońska, Z. Ciunik and C. Wawrzeńczyk, Tetrahedron: Asymmetry, 2005, 16, 2087–2097 CrossRef CAS.
- P. W. Wellings, S. A. Ward, A. F. G. Dixon and R. Rabbinge, in Aphids, Their Biology, Natural Enemies and Control, ed. A. K. Minks and P. Harrewijn, Elsevier Amsterdam, 1989, vol. C, pp. 49–64 Search PubMed.
- N. I. Katis, J. A. Tsitsipis, M. Stevens and G. Powell, in Aphids as Crop Pests, ed. H. F. v. Emden and R. Harrington, CABI, Wallingford, 2007, pp. 353–390 Search PubMed.
- C.-A. Dedryver, A. Le Ralec and F. Fabre, C. R. Biol., 2010, 333, 539–553 CrossRef.
- E. Prado and W. F. Tjallingii, Entomol. Exp. Appl., 1994, 72, 157–165 CrossRef.
- G. Powell, T. Pirone and J. Hardie, Eur. J. Plant Pathol., 1995, 101, 411–420 CrossRef.
- B. Martin, J. Collar, W. Tjallingii and A. Fereres, J. Gen. Virol., 1997, 78, 2701–2705 CAS.
- V. Brault, M. Uzest, B. Monsion, E. Jacquot and S. Blanc, C. R. Biol., 2010, 333, 524–538 CrossRef.
- R. L. Blackman and V. F. Eastop, Aphids on the World's Crops: An Identification and Information Guide, John Wiley and Sons, Chichester, 1985 Search PubMed.
- J. A. Pickett, L. J. Wadhams and C. M. Woodcock, Brighton Crop Protection Conference – Pests and Diseases, British Crop Protection Council, Thornton Heath, UK, 1994, pp. 1239–1246 Search PubMed.
- K. Wińska, A. Grudniewska, A. Chojnacka, A. Bialońska and C. Wawrzeńczyk, Tetrahedron: Asymmetry, 2010, 21, 670–678 CrossRef.
- W. S. Johnson, L. Werthemann, W. R. Bartlett, T. J. Brocksom, T. T. Li, D. J. Faulkner and M. R. Petersen, J. Am. Chem. Soc., 1970, 92, 741–743 CrossRef CAS.
- K.-K. Chan, A. C. Specian and G. Saucy, J. Org. Chem., 1978, 43, 3435–3440 CrossRef CAS.
- A. Srikrishna and C. Dinesh, Tetrahedron: Asymmetry, 2005, 16, 2203–2207 CrossRef CAS.
- A. Srikrishna, S. Nagaraju and P. Kondaiah, Tetrahedron, 1995, 51, 1809–1816 CrossRef CAS.
- N. Yoshikawa, L. Tan, N. Yasuda, R. P. Volante and R. D. Tillyer, Tetrahedron Lett., 2004, 45, 7261–7264 CrossRef CAS.
- M. Hiersemann and U. Nubbemeyer, The Claisen Rearrangement. Methods and Applications, WILEY-VCH Verlag GmbH & Co. KGaA Weinheim, 2007 Search PubMed.
- K. Mori and Y. Nakazono, Tetrahedron, 1986, 42, 283–290 CrossRef CAS.
- G. Cardillo and M. Orena, Tetrahedron, 1990, 46, 3321–3408 CrossRef CAS.
- M. D. Dowle and D. I. Davies, Chem. Soc. Rev., 1979, 8, 171–197 RSC.
- S. Fuchs, V. Berl and J. P. Lepoittevin, Eur. J. Org. Chem., 2007, 1145–1152 CrossRef CAS.
- S. Ranganathan, K. M. Muraleedharan, N. K. Vaish and N. Jayaraman, Tetrahedron, 2004, 60, 5273–5308 CrossRef CAS.
- B. B. Snider and M. I. Johnston, Tetrahedron Lett., 1985, 26, 5497–5500 CrossRef CAS.
- J. L. Frazier and S. Chyb, in Regulatory Mechanisms in Insect Feeding, ed. R. F. Chapman and G. d. Boer, Chapman & Hall, New York, 1995, pp. 364–381 Search PubMed.
- Oxford Diffraction. CrysAlis ‘RED’, Wrocław, Oxford Diffraction (Poland) Sp. z o.o., 2001, 2003 Search PubMed.
- SHELXTL-NT, v. 5.1, Bruker AXS, 1999 Search PubMed.
- P. Harrewijn, in Aphid - Plant Genotype Interactions, ed. R. K. Campbell and R. D. Eikenbarry, Elsevier, Amsterdam, 1990, pp. 117–130 Search PubMed.
- J. Hardie, M. Holyoak, N. J. Taylor and D. C. Griffiths, Entomol. Exp. Appl., 1992, 62, 233–239 CrossRef.
- W. F. Tjallingii, in Regulatory Mechanisms in Insect Feeding, ed. R. F. Chapman and G. d. Boer, Chapman & Hall, New York, 1995, pp. 190–209 Search PubMed.
- W. F. Tjallingii, Eur. J. Entomol., 1994, 91, 47–52 Search PubMed.
- M. Mayoral, W. F. Tjallingii and P. Castañera, Entomol. Exp. Appl., 1996, 78, 341–348 CrossRef.
- M. Mazur, W. Gładkowski and C. Wawrzeńczyk, Przem. Chem., 2011, 90, 918–922 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra (1H NMR, COSY, 13C NMR, HSQC) of compounds 2c, 3c, 4c, 5c, 7c, 8c, 9c, 10c, 11c and CGC chromatograms of compounds 4a–c, 5a–c, 9a–c, 10a–c, 11a–c. CCDC reference numbers 825643–825650. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1ra00270h |
| ‡ This paper is a part 37 of series Lactones.60 |
| This journal is © The Royal Society of Chemistry 2011 |