Photochemical behavior of PVA as an oxygen-barrier polymer for solar cell encapsulation
Julien
Gaume
ab,
Pascal
Wong-Wah-Chung
bc,
Agnès
Rivaton
ab,
Sandrine
Thérias†
*ab and
Jean-Luc
Gardette
ab
aClermont Université, Université Blaise Pascal, LPMM, BP 10448, F-63000 Clermont-Ferrand, France. E-mail: sandrine.therias@univ-bpclermont.fr; Fax: +33 (0)4 73 40 77 00; Tel: +33 (0)4 73 40 71 43
bCNRS, UMR 6505, LPMM, BP 80026, F-63171 Aubiere, France
cClermont Université, ENSCCF, LPMM, BP 10448, F-63000 Clermont-Ferrand, France
First published on 12th October 2011
Abstract
Polyvinyl alcohol (PVA) is a water-soluble polymer that is anticipated to be a good candidate for incorporation into multilayer coatings of organic solar cells due to its high transparency and ability to form a barrier to oxygen. Because a long lifetime is a prerequisite for successful applications, it was necessary to study the photochemical behavior of PVA under solar light. PVA films were exposed to UV-visible light irradiation (λ > 300 nm) in accelerated aging conditions representative of natural ageing. Modifications in the chemical structure of aged samples irradiated at ambient air were recorded. Due to the low oxygen permeability of PVA films, it was shown that the photooxidative degradation of PVA films is restricted to the surface (<5 μm) and results in a large amount of chain scissions, with a progressive erosion of the surface of the irradiated material. The oxidation products formed along the macromolecular chains, and low molecular weight species trapped in the matrix or emitted in the gas phase were also identified. An oxidation mechanism was then proposed to account for these modifications. However, irradiation in the absence of oxygen demonstrated the high photostability of PVA films, which permits the use of PVA as a sublayer in inorganic/organic multilayer encapsulation systems.
1. Introduction
Polyvinyl alcohol (PVA) is extensively used in paper coating, textile sizing, and in packaging as flexible water-soluble films1 for its oxygen barrier effect. PVA is also used in numerous fields because of its biodegradability and environmentally friendly processing.2,3 Indeed, PVA is a water-soluble polymer that permits the formation of transparent films through the evaporation of an aqueous solution.It is well known that organic solar cells (OSCs) are sensitive to the oxygen and water present in the atmosphere and therefore require encapsulation by efficient barriers.4–6 To obtain flexible organic solar cells with long lifetimes, it appears necessary to develop encapsulation with a large range of properties: a high barrier to water and oxygen, optical transparency in the visible domain, flexibility and environmentally friendly processing. Moreover, all these properties have to be preserved in use conditions under sunlight exposure (λ > 300 nm). Regarding the high barrier properties to oxygen4,5 (PO2PVA = 3 × 10−17 cm3 cm/cm2 s Pa compared to PO2PET = 3 × 10−15 cm3 cm/cm2 s Pa) and the transparency of PVA, this polymer was anticipated to be a good candidate for incorporation into a multilayer inorganic/organic system for organic solar cell (OSC) encapsulation.6–9 The water barrier property would be provided by the alternating inorganic layers. However, the photochemical behavior of pure PVA must be studied before its integration into an inorganic/organic multilayer structure.
To our knowledge, no study has focused on the photodegradation of PVA under UV-visible light irradiation in conditions representative of outdoor exposure (λ > 300 nm). Some papers report on the phototransformation of PVA irradiated at short wavelength (254 nm) in the presence of collagen,10,11 montmorillonite12 or graphite oxide.13 Another paper focused on the photochemical behavior of dichromated PVA upon exposure at 365 nm.14 On the basis of published results, it seems that PVA could be considered a photostable polymer and could be a good candidate in multilayer encapsulation systems for OSCs with long lifetimes.
The present study focuses on the photochemical behavior of PVA and on the modifications of the chemical structure of PVA resulting from photo-thermal ageing. PVA thin films (20–30 μm thick) were submitted to UV-visible light irradiation (λ > 300 nm, 60 °C) in ambient air or were thermally oxidized at 60 °C. Chemical modifications were investigated by X-ray photoelectron spectroscopy XPS, IR and UV-visible spectroscopies. The photoproducts were identified by various techniques combining physical and chemical derivatization treatments of oxidized films coupled with IR spectroscopy. Identification of the low molecular weight products was performed by head-space solid-phase micro-extraction (HS-SPME)-coupled with GC-MS analysis and ionic chromatography experiments. The obtained data allowed us to propose a detailed degradation mechanism that accounted for the formation of the major oxidative species. Depth profiling was used to determine the extent of the oxidative process throughout the polymer films. Lastly, the degradative changes of PVA submitted to irradiation in the absence of oxygen was also analyzed. The results indicated the main parameters that must be managed for PVA to serve as a good candidate for insertion in a multilayer system for OSC encapsulation.
2. Experimental
2.1. Materials
It is important to note that PVA is obtained by the hydrolysis of polyvinyl acetate. Usually, the reaction is not complete, and acetate functions can remain in the polymeric structure. Consequently, PVA with different hydrolysis percentages in the range 70–99% are commercially produced.In this study, PVA (98% hydrolyzed, weight-average molecular weight ≈ 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) (Fig. 1) was purchased from Sp2 (Scientific Polymer Products). Before use, the PVA was purified with methanol in a Soxhlet apparatus for two days.
000 g mol−1) (Fig. 1) was purchased from Sp2 (Scientific Polymer Products). Before use, the PVA was purified with methanol in a Soxhlet apparatus for two days.
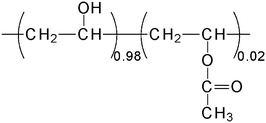 | ||
| Fig. 1 Chemical structure of PVA. | ||
The purified PVA was then dissolved in water (5 wt%) at 80 °C under stirring for 3 h. Then the PVA aqueous solution was deposited on a Teflon sheet with an Erichsen Coatmaster 809 MC (thickness of the liquid deposition ∼ 300 μm). Free-standing films with thicknesses in the range 20–30 μm were obtained after drying in ambient air for one day plus a few hours at 60 °C.
2.2. UV-visible irradiation
The UV-visible light irradiation (λ > 300 nm) of the thin films was performed in a SEPAP 12/24 unit, which was designed for studying polymer photodegradation in artificial ageing with medium-accelerated conditions.15 The chamber consisted of a square reactor equipped with four medium-pressure mercury lamps (Novalamp RVC 400W) situated vertically at each corner of the chamber. Wavelengths below 300 nm are filtered by the glass envelope of the lamps. In the center of the chamber, the samples are fixed on a 13 cm-diameter rotating carousel that could hold 24 samples. In this set of experiments, the temperature at the surface of the samples was set at 60 °C. Irradiation in the absence of oxygen (photolysis experiments) was performed on samples introduced into borosilicate tubes and sealed under a vacuum of 10−4 Pa with a diffusion vacuum line. The samples in tubes under vacuum were then placed in the SEPAP 12/24 device.Low-temperature thermooxidation experiments were carried out in a ventilated oven at 60 °C.
2.3. Characterization
Changes in the UV-visible spectra were tracked with a Shimadzu UV-2101PC spectrophotometer equipped with an integrating sphere.IR spectra were recorded in transmission mode with a Nicolet 6700 Fourier transform infrared (FTIR) spectrophotometer operated with the OMNIC software. The spectra were obtained with 32 scan summations at 4 cm−1 resolution. IR-ATR (Attenuated Total Reflectance) spectra were recorded in the reflection mode with a Nicolet 380-FTIR spectrophotometer equipped with a Thunderdome-ATR (4 cm−1, 32 scans). The Thunderdome is a single reflection ATR accessory with a germanium crystal (depth analysis ≈ 1 μm). Oxidation profiles were determined by recording spectra in the IR-ATR mode after successive abrasions of the film surface.
Most of the oxidation products were identified by performing chemical derivatization treatments that selectively convert oxidation products into chemical groups with different IR characteristics.16,17Ammonia (NH3) reacts with carboxylic acids to generate carboxylate ions. NH3 also reacts with esters to give amides. NH3 treatment was performed at room temperature in simple flow reactors that could be sealed off to allow the reaction to proceed. Irradiated films were also treated with a solution of 2,4-dinitrophenylhydrazine (DNPH) in methanol for 18 h. DNPH reacts with aldehydes and ketone groups to give dinitrophenylhydrazones. Before DNPH treatment, the samples have to be first immersed in methanol solution to eliminate low molecular weight oxidation products.
X-Rray photoelectron spectroscopy (XPS) experiments were conducted in a PHI Quantera SXM. The diameter of the analyzed area was 200 μm, and the depth of analysis was less than 10 nm.
Ionic chromatography analyses (Dionex AS11 column for anions, eluant NaOH from 0.5 mmol L−1 to 25 mmol L−1, injection 100 μL at 30 °C) were performed in a water immersion solution of a photooxidized PVA film.
Films of PVA were also irradiated in sealed vials to collect the volatile photodegradation products. Carboxen–PDMS fiber (75 μm) purchased by Supelco (Bellefonte, PA, USA) was used to extract the volatile products. The extraction time was 5 min at 80 °C. The volatile compounds were analyzed by gas chromatography/mass spectrometry (GC-MS) analysis with a 6890N Agilent GC coupled to a 5973 Agilent mass detector. The GC was equipped with a Supelcowax™ 10 column (30 m × 0.25 mm × 0.25 μm) from Supelco. Splitless injections were used (2 min). The temperature was programmed from 35 °C (10 min hold) to 60 °C at 5 °C min−1 and then at 10 °C min−1 to 200 °C (15 min hold). Helium was used as a carrier gas with a constant pressure of 38 kPa. The temperature of the splitless injector was 280 °C (2 min duration), and the flow was 50 mL min−1. The transfer line temperature was 280 °C, and the ion source temperature was kept at 230 °C. The ionization occurred with a kinetic energy of the impacting electrons of 70 eV. The detector voltage was 70 eV. Mass spectra and reconstructed chromatograms (total ion current, TIC) were acquired under the electron ionization mode (EI) at 70 eV and recorded from 20 to 400 m/z. The compounds were identified either by comparison of the retention times and mass spectra with standards or with the spectral library.
3. Results and discussion
3.1. Thermooxidation of PVA at 60 °C
Thermal degradation of PVA has been reported in the literature at a high temperature (T > 200 °C),18 which is significantly higher than those expected for OSCs (<80 °C). It was shown that the thermal degradation involves the elimination of hydroxyl side groups together with the formation of isolated and conjugated polyenes and carbonyl groups. To discriminate the influence of temperature and light on the oxidation provoked by exposure to UV light in the artificial ageing unit, thermooxidation experiments were performed in a ventilated oven at 60 °C. After 7000 h of thermooxidation, no significant change of the IR spectra was observed. The only modifications concerned the UV-visible spectra (Fig. 2), which showed an absorption band at 280 nm and a shoulder at 330 nm, which were assigned to the π–π* transitions of –(CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH)2–CO– and –(CHCH)3–CO–, respectively.19,20
CH)2–CO– and –(CHCH)3–CO–, respectively.19,20
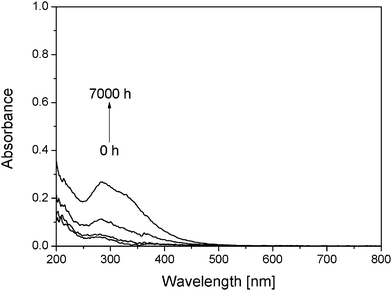 | ||
| Fig. 2 UV-Visible spectra of a PVA thin film during thermooxidation at 60 °C. | ||
The products that resulted from oxidation and dehydration of PVA were formed at low concentrations and were only observed by UV-visible analysis because the extinction coefficient of the involved electronic transitions is important. As a consequence of the weak concentrations, no modification of IR spectra corresponding to these products was observed.
3.2. Photolysis of PVA at long wavelengths (λ > 300 nm)
The irradiation of the PVA was performed in the absence of oxygen (photolysis) at 60 °C. After 6 000 h in the SEPAP 12/24 unit, only a fairly weak dehydration (elimination of absorbed water) of the samples was observed. Otherwise, no significant changes of the UV-visible and IR spectra in the chemical structure were observed.3.3. Photooxidation of PVA at long wavelengths (λ > 300 nm)
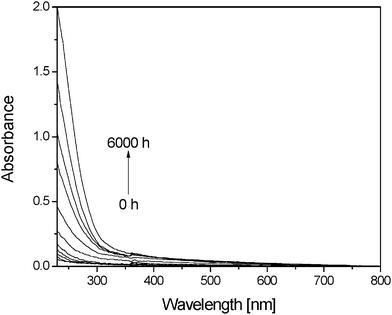 | ||
| Fig. 3 UV-visible spectra of a PVA thin film during photooxidation in the SEPAP 12/24 device at λ > 300 nm and 60 °C | ||
Fig. 3 also shows that photooxidation of PVA films resulted in a gradual increase of light absorption from 300 to 500 nm without any maximum. The increase in absorbance at 400 nm after 6000 h of exposure was not higher than 0.06, which indicates that no significant discoloration was observed. In comparison to thermooxidation experiments, no absorption bands corresponding to the formation of polyenes were detected. Although conjugated products could be formed, they would be rapidly oxidized under irradiation and then disappear, as observed in the case of poly(vinyl chloride).21,22
3.3.2.1. Infrared analysis of the chemical changes in the polymer matrix. Modifications in the bulk of PVA films were analyzed by transmittance-IR spectroscopy after 6000 h of irradiation. The analysis of changes in IR spectra requires that the absorption bands composing the spectrum of native PVA are correctly identified. The main spectral features are presented in Table 1.14,23–25
| Wavenumbers (cm−1) | Assignments |
|---|---|
| 3340 | OH stretching |
| 2942 | CH2 stretching |
| 2914 | C–H stretching |
| 2853 | C–H stretching |
| 1730 | CO stretching of free ester groups |
| 1713 | CO stretching of ester groups linked by H bonds to hydroxyl groups of PVA |
| 1420 | CH2 bending |
| 1377 | CH3 wagging |
| 1246 | C–O–C stretching |
| 1096 | O–H bending |
| 918 | CH2 rocking |
| 853 | C–C stretching |
Photooxidation resulted in noticeable modifications in the IR spectrum of the PVA (Fig. 4), which can be observed in the subtracted spectra.
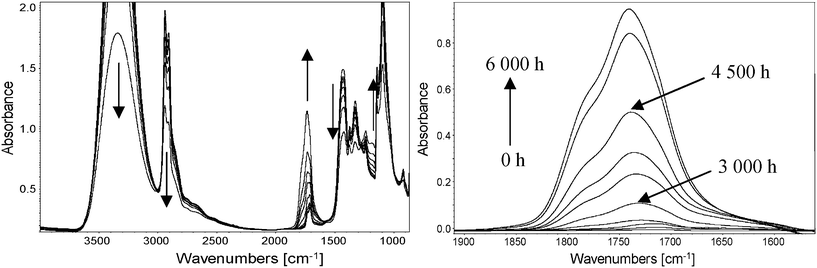 | ||
| Fig. 4 (left) Direct IR spectra of a PVA film photooxidized at λ > 300 nm and 60 °C in the SEPAP 12/24 device for 6000 h. (right) Subtracted spectra in the carbonyl region. | ||
The vibration bands corresponding to –OH groups (3340 cm−1) and the (CH) polymer backbone (2940 and 2910 cm−1) strongly decreased in intensity upon irradiation. In the carbonyl region, one can observe the formation of a complex envelope containing a maximum at 1740 cm−1 and a shoulder at around 1785 cm−1 in the subtracted spectra. The formation of a band with a maximum at approximately 1180 cm−1 was also observed.
3.3.2.2. Identification of oxidation products by chemical derivatization treatments. To identify the oxidation products detected by IR analysis, which are formed on the macromolecular chains or trapped in the bulk, chemical derivatization treatments were performed on photooxidized PVA samples. The combination of IR data with derivatization treatments is known to be particularly efficient for the identification of carbonyl compounds.17
NH3 reacts with carboxylic acids to generate carboxylate ions and with esters, anhydrides and lactones to generate amides. It was first verified that no reaction with NH3 was observed with PVA before photooxidation. Fig. 5 shows the subtracted spectra of a PVA sample exposed for 5000 h compared to a non-irradiated sample before and after NH3 treatment. The IR spectrum monitored after treatment indicates the formation of carboxylate (major) and amide (minor) groups, with the concomitant disappearance of oxidation photoproducts centered at 1785 cm−1, 1740 cm−1 and 1715 cm−1.
On the basis of the numerous publications concerning derivatizations by NH3,17 the oxidation photoproduct observed at 1715 cm−1, which reacted with NH3 to give the carboxylate group observed at 1570 cm−1, was assigned to a carboxylic acid in the dimer form. The formation of an amide group (shoulder at 1670 cm−1) can be assigned to the NH3 reaction with anhydrides or lactones observed at 1785 cm−1. In the case of the oxidation photoproduct observed at 1740 cm−1, this frequency is usually associated with ester functions, which react with NH3 to give amide groups. In the case of PVA, this absorption band could also reflect the formation of α-hydroxylated carboxylic acid. To check this hypothesis, a carboxylic acid (propanoic acid CH3CH2COOH) and an α-hydroxylated carboxylic acid (lactic acid CH3CHOHCOOH) were introduced in two different samples of native PVA film that were then treated with NH3. Table 2 gives the absorption maxima of propanoic acid and lactic acid in PVA and the absorption bands of the derivative products after NH3 treatment.
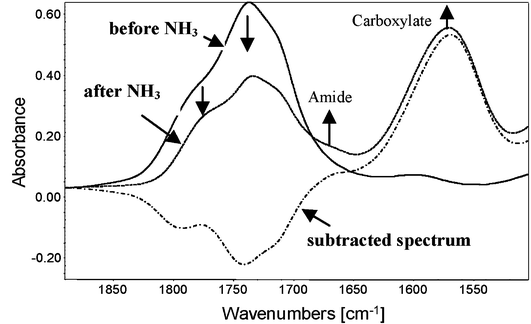 | ||
| Fig. 5 Subtracted spectra of photooxidized PVA (5000 h) (compared to non-irradiated PVA) before and after NH3 treatment and the subtracted spectra (after—before NH3 treatment). | ||
| Wavenumber (cm−1) | |||
|---|---|---|---|
| Acid | Pure | Acid in PVA | Derived product |
| Propanoic | 1713 | 1713 | 1565 |
| Lactic | 1730 | 1745 | 1600 |
One can see that the presence of a –OH group in the α-hydroxylated carboxylic acid (lactic acid) resulted in a shift (Δυ = 17 cm−1) of the vibration band attributed with the pure α-hydroxylated carboxylic acid. When these acids were introduced in PVA, the shift was even more significant (Δυ = 28 cm−1) due to hydrogen bonds. Carboxylate derived products also gave a shift (Δυ= 35 cm−1) after NH3 treatment. Based on these results, it can be assumed that the oxidation products observed at 1740 cm−1 in the photooxidized PVA film could be assigned to ester groups and α-hydroxylated carboxylic acids.
After NH3 treatment, some unreacted oxidation products remain in the PVA film. A 2,4-DNPH treatment was conducted on irradiated PVA films, previously immersed in methanolic solution (to extract the low molecular weight species) and then soaked in a methanolic solution of 2,4-DNPH for 18 h. It is important to note that 2,4-DNPH treatment gives qualitative information about the presence of ketone and aldehyde.26,27 The reaction with 2,4-DNPH (Fig. 6) led to a decrease in intensity of the 1717 cm−1 absorption band (before the treatment with DNPH, it was verified that the decrease of this band did not result from the immersion of the irradiated films in the solvent only). At the same time, two bands at 1618 and 1592 cm−1 appeared, which can be assigned to the corresponding phenylhydrazone.
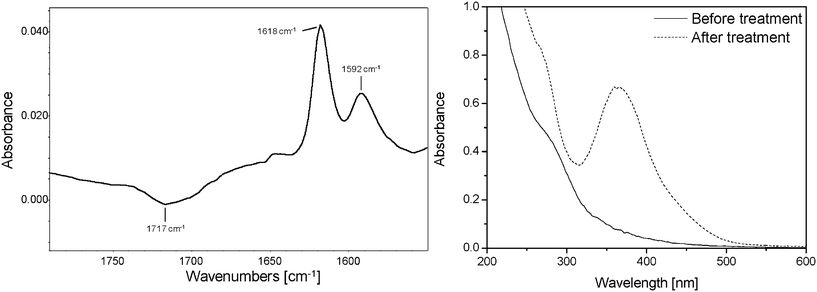 | ||
| Fig. 6 (Left) IR difference spectra (after—before) the 2,4-DNPH treatment. (Right) UV-visible spectra (after—before) the 2,4-DNPH treatment. | ||
The formation of phenylhydrazone could also be followed by UV-visible spectroscopy with the appearance of an intense absorption at 365 nm (Fig. 6). To identify the chemical nature (ketone or aldehyde) of the product reacting with 2,4-DNPH, a physical treatment was performed on a photooxidized PVA film. This film was placed above water vapor to eliminate low molecular weight acid groups and then submitted to a photolysis experiment (irradiation in the absence of oxygen). The subtracted difference spectrum (after—before photolysis) revealed a decrease in the vibration band at 1717 cm−1 upon photolysis. Due to the well-known photolytic instability of ketone according to the Norrish reaction, the band at 1717 cm−1 was attributed to a ketone. Ketone functions are generally detected at approximately 1720 cm−1. However, it is well-known that hydrogen bonding between carbonyl and hydroxyl groups shifts the CO stretching vibrations to lower wavenumbers.14
3.3.2.3. Identification of oxidation products by ionic chromatography. A PVA film photooxidized for 4000 h was immersed in 2 mL of water during 2 min. The film was analyzed with IR spectroscopy before and after immersion, and the subtracted spectra to native PVA are reported in Fig. 7.
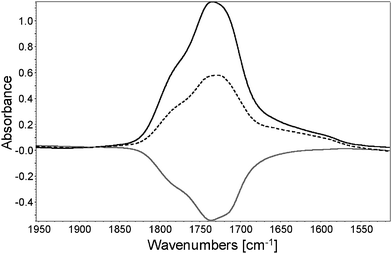 | ||
| Fig. 7 Subtracted spectra (after exposure—t0) (black line), (after water immersion—t0) (dotted line) and (after—before) water immersion (grey line) | ||
The immersion of photooxidized PVA in water leads to a 45% decrease of the IR absorption of the carbonyl envelope, which indicates the extraction of some low molecular weight products trapped in the film. As observed by NH3 treatment, the subtracted spectrum shows the decrease of three maxima centered at 1785 cm−1, 1740 cm−1 and 1715 cm−1. To identify the extracted products, the water solution was analyzed with ionic chromatography (results reported in Table 3). By comparison with standards, the primary released products were identified as acetic, oxalic, malonic and succinic acids. α-Hydroxylated carboxylic acids such as lactic and tartaric acids were also present in minor proportions.
| Compound | Formula | Molecular weight (g mol−1) | C (μg L−1) | C (μM) |
|---|---|---|---|---|
| Formic acid | H–COOH | 46.03 | 220 | 4.78 |
| Acetic acid | H3C–COOH | 60.05 | 3560 | 59.28 |
| Propionic acid | H3C–CH2–COOH | 74.08 | 150 | 2.02 |
| Oxalic acid | HOOC–COOH | 90.03 | 2140 | 23.77 |
| Malonic acid | HOOC–CH2–COOH | 104.06 | 1890 | 18.16 |
| Succinic acid | HOOC–CH2–CH2–COOH | 118.09 | 1610 | 13.63 |
| Lactic acid | H3C–CHOH–COOH | 90.08 | 300 | 3.33 |
| Tartaric acid | HOOC–CHOH–CHOH–COOH | 150.09 | 670 | 4.46 |
3.3.2.4. XPS analysis. The analysis of the bulk oxidation with IR spectroscopy shows weak photoproduct concentrations after 2500 h. The modifications detected by IR analysis in transmission mode correspond to the mean concentrations of photoproducts within the PVA films. Due to the high oxygen barrier of PVA, it can be anticipated that most of the oxidation photoproducts are localized at the surfaces of the irradiated films. The chemical modifications of the surface after 2500 h of exposure were studied with XPS. XPS is commonly accepted as one of the most powerful tools to monitor chemical changes in polymer surfaces (to a depth of approximately 10 nm). The investigation of the core electronic structures of the elements gives precise information about the chemical environment of the different atoms. An XPS spectrum of a PVA sample was recorded. As expected, carbon (C1s at 285 eV) and oxygen (O1s at 532 eV) atoms were found as the two major constituents. Using the C1s and the O1s peak areas, the oxygen/carbon atomic ratio (O/C) was found to be 0.47, which is in good agreement with the ideal stoichiometry of PVA (0.50). The C1sspectrum consisted of four subpeaks at 285.0, 286.4, 287.6 and 289.1 eV (Fig. 8). The lowest binding energy was assigned to C–C/C–H bonds, while the subpeak at 286.4 eV was assigned to C–O bonds (alcohol functions). The two weaker subpeaks at 287.6 and 289.1 eV were assigned to C
O and O–CO bonds, respectively, originating from residual acetate groups present in PVA.
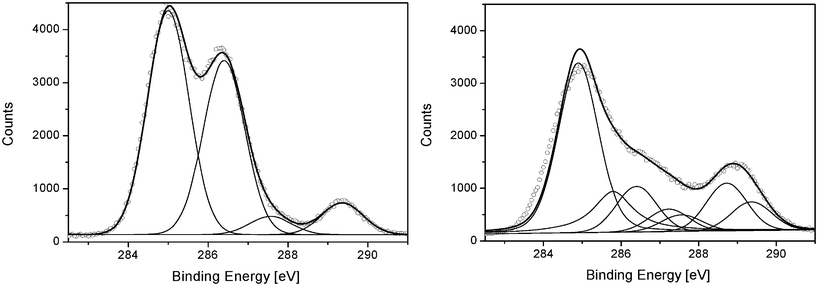 | ||
| Fig. 8 C1s fitted curves of pristine PVA (left) and after 2500 h of photooxidation (right). | ||
After 2500 h of exposure, modifications to the XPS spectrum were observed. A significant increase of the O/C ratio (0.57) due to oxygen fixation and the formation of oxidized products was observed. The C1sspectrum of the photooxidized sample was resolved using the same subpeaks as those for an un-irradiated PVA and adding subpeaks corresponding to the formation of oxidation products. Table 4 summarizes the C1s deconvolution results before and after irradiation.
| BE (eV) | Assignment | 0 h | 2500 h |
|---|---|---|---|
| 285 | C–C and C–H | 49.9 | 45.5 |
| 286.4 | C–O | 38.9 | 11.2 |
| 287.6 | CO |
4.1 | 4.1 |
| 289.1 | O–CO |
7.1 | 7.1 |
| 285.9 | C–O–C | — | 14.8 |
| 287.2 | CO |
— | 5.5 |
| 288.7 | O–CO |
— | 11.8 |
Photooxidation led to the decrease of C–C and C–H content and the decrease of C–O bonds. This suggests that the oxidation of the alkyl chains and the chain scission of C–O bonds with the elimination of alcohol function occur simultaneously upon exposure. The peaks situated at 287.6 and 289.1 eV were assigned to the CO and O–CO of acetate groups present in PVA and are not affected by photooxidation. As previously suggested by Akhter et al.,28 a peak at 285.9 eV must be added to correctly fit the data. This supplementary peak was tentatively assigned to ether C–O–C functions induced by radical recombination. The formations of the subpeaks at 287.2 and 288.7 eV correspond to the formation of carbonyl products. Although few products were detected in the bulk, the surface was already oxidized after 2500 h of exposure. This confirms, therefore, that the oxidation process leads to the lower intensity of the C–H/C–C subpeak at 285 eV. The first subpeak (287.2 eV) was assigned to ketone formation (CO), whereas the second subpeak (288.7 eV) was assigned to carboxylic acids formation (O–CO).
3.3.2.5. Oxidation photoproducts concentrations. The oxidation products assigned to the increase in IR absorbance at 1715 cm−1, 1717 cm−1, 1740 cm−1 and 1785 cm−1 after photooxidation of PVA are listed in Table 5.
| Wavenumber (cm−1) | Oxidation products | Identification |
|---|---|---|
| 1715 | Carboxylic acids | NH3 treatment, ionic chromatography |
| 1717 | Ketones | 2,4-DNPH treatment and photolysis |
| 1740 | Esters and carboxylic acids | NH3 treatment, ionic chromatography |
| 1785 | Lactones and anhydrides | NH3 treatment |
Based on these results, it should be possible to compare the concentration of carbonyl products present in the PVA film and detected by IR analysis to the (C–H) consumption. Clearly, the oxidation reaction of polymers involves a hydrogen abstraction by a radical process leading to the loss of C–H bonds with the formation of CO bonds.29 The carbonyl envelope developed in the IR spectra upon irradiation was deconvoluted with three contributions. The first contribution was fixed at 1715 cm−1 and was attributed to carboxylic acids and ketones. The second contribution was fixed at 1745 cm−1 and was attributed to ester and carboxylic acids, whereas the third contribution was fixed at 1785 cm−1 and was attributed to lactones and anhydrides. Each contribution was represented by a Gaussian curve with a fixed width at half height (FWHM = 47 cm−1). To obtain the (C–H) concentration, the decrease of the vibration band at 2942 cm−1 was measured. The average concentrations were determined using the Beer–Lambert law (Abs = εcl). The molar extinction coefficients depend on the nature of the products. Extinction coefficients of some model carbonyl compounds, as measured by Carlsson and Wiles,30 range from 350 L mol−1 cm−1 for ketones to 650 L mol−1 cm−1 for acids. We have shown that the absorption bands can result from two different products at the same time (acids and ketones at 1715 cm−1, acids and esters at 1745 cm−1). For this reason, we used an average value for the extinction coefficient of these bands. The concentrations of the products at 1715 cm−1 were then calculated with a coefficient of 500 L mol−1 cm−1, and the concentration of the products at 1745 cm−1 were calculated with a coefficient of 550 L mol−1 cm−1. In the case of the products at 1785 cm−1, we used a coefficient of 450 L mol−1 cm−1. On the basis of the infrared spectra of several different polymer films with a known thickness, the extinction coefficient of the (CH) groups at 2940 cm−1 was evaluated to be approximately 10 L mol−1 cm−1.
Fig. 9 shows an example of the fit for a PVA film after 5600 h of exposure and the kinetics of carbonyl products formation and (C–H) consumption in the PVA film upon irradiation.
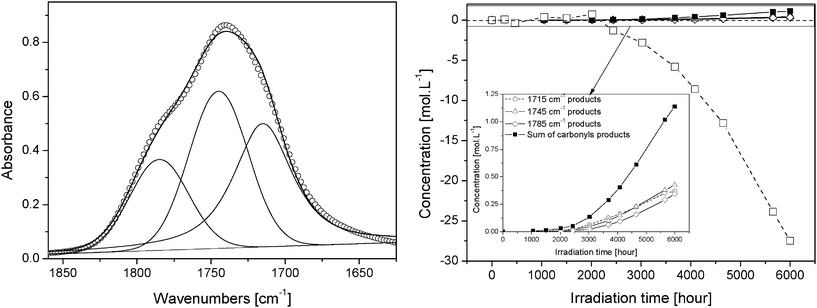 | ||
| Fig. 9 (Left) Fitted curves of subtracted carbonyl envelop after 5600 h of exposure. (Right) Kinetics of carbonyl products formation (■) and (C–H) consumption (□) in PVA film during photooxidation. | ||
One can observe that the loss of (CH) groups is significantly higher than the accumulation of (CO) groups in PVA irradiated films. For instance, after 6000 h of irradiation, a C–H concentration decrease of about 25 mol L−1 was obtained, whereas the concentration of oxidation products formed in the PVA film was less than 1.5 mol L−1. These results indicate that some of the oxidation products that were formed are not taken into account by the IR analysis of the irradiated PVA film. This result suggests the formation of low molecular weight species susceptible to migration in the gas phase upon irradiation, as reported for other polymers.31,32
3.3.2.6. Identification of volatile products: SPME. To identify the volatile photoproducts, HS-SPME-GC-MS experiments were performed. The GC-MS chromatogram (TIC), shown in Fig. 10, shows that various low molecular weight products have migrated in the gas phase after 4000 h of exposure.
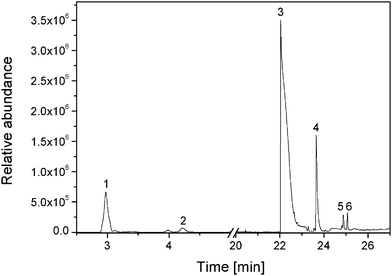 | ||
| Fig. 10 HS-SPME-GC-MS total ion chromatogram of a headspace of a PVA film after 4000 h exposure. | ||
The retention times, tret, the main observed fragments, the molecular weights of the compounds and the identification of the products were given in Table 6.
| Number | tret (min) | Main fragment, m/z | Molecular weight (g mol−1) | Identification |
|---|---|---|---|---|
| 1 | 2.9 | 58, 43, 27 | 58.1 | Acetone |
| 2 | 3.4 | 72, 57, 43, 29 | 72.1 | Butanone |
| 3 | 22.1 | 60, 43, 29 | 60.0 | Acetic acid |
| 4 | 23.6 | 101, 75, 57, 41, 29 | 74.1 | Propanoic acid |
| 5 | 24.8 | 88, 73, 60, 42 | 88.1 | Butanoic acid |
| 6 | 25.1 | 86, 56, 42, 29 | 86.1 | Butyrolactone |
According to the total ion abundance, the results given in Table 6 indicate that the products with retention times of 2.9, 22.1 and 23.6 min were the main products detected during the photooxidation process. These three volatile products were identified as acetone, acetic acid and propanoic acid, respectively. Other products, such as butanone, butanoic acid or butyrolactone, were detected in lower quantities.
O band is accompanied by a dramatic decrease of the absorbance of the C–H bands at 2942 cm−1 and 2914 cm−1. This indicates that the oxidation photoproducts were mainly formed at the surface of the exposed samples.
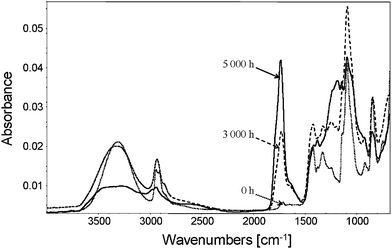 | ||
| Fig. 11 IR-ATR spectra of a pristine PVA and oxidized PVA (3000 h and 5000 h). | ||
To determine the distribution of the oxidation products within the sample thickness, the IR-ATR spectra were recorded after successive abrasions of the surface. The ratio oxidation products/C–H are presented in Fig. 12.
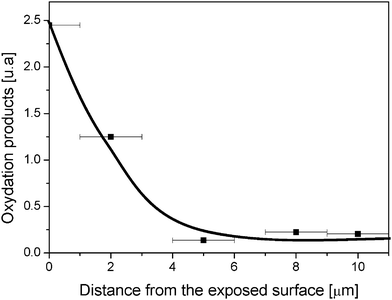 | ||
| Fig. 12 Photooxidation profile (CO/CH ratio) measured by IR-ATR spectroscopy after successive abrasions of a PVA film irradiated for 3000 h at λ > 300 nm and 60 °C. | ||
The shape of the profile shows a marked concentration gradient. The oxidation photoproducts were formed at the surface of the film within a layer that is less than 5 μm from the exposed surface. This profile reflects the high barrier of PVA to oxygen,4 which limits the oxidation processes in the bulk of PVA.
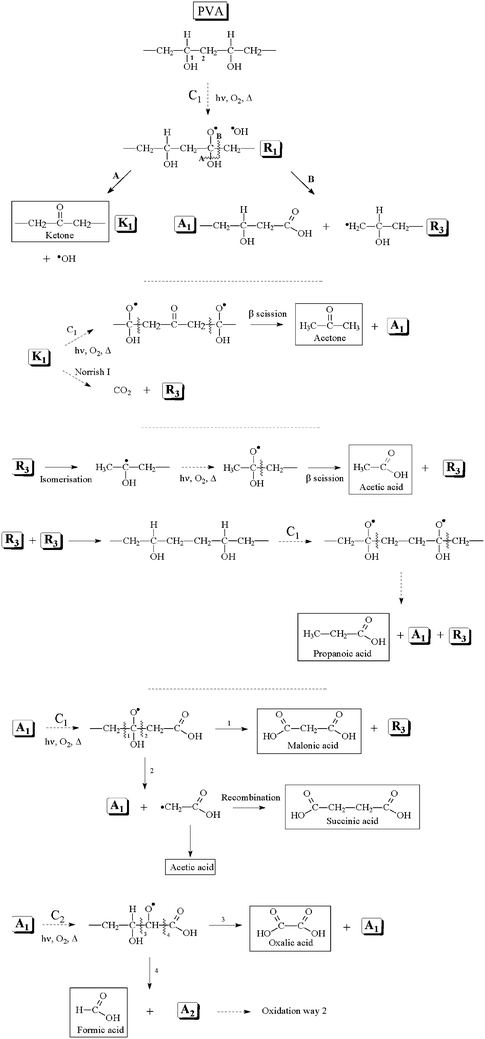 | ||
| Scheme 1 Oxidation mechanism of the PVA with a primary attack on the tertiary carbon (C1). | ||
It is well known that the hydrogen atom on a tertiary carbon atom is a preferential site for a radical attack. Such behavior has been clearly demonstrated in the degradation mechanism of numerous polymers, including common polymers such as polyvinyl chloride21 or polypropylene.33Hydrogen abstraction at the tertiary carbon atom (C1 in Scheme 1) leads to the formation of a macroalkyl radical, and after subsequent oxygen fixation and hydrogen abstraction, hydroperoxides are formed as primary oxidation products. The photochemical decomposition of the hydroperoxides leads to alkoxy radicals (R1). A β-scission of the C–O bond (A) on R1 may occur, forming a chain ketone (K1) and a OH• radical. A β-scission of C–C bond (B) on R1 also occurs and forms a carboxylic acid (A1) and an alkyl primary macroradical (R3). K1, R3 and A1 are formed as the secondary products. The formation of acetone can be attributed to the oxidation of K1, which involves two successive oxidations on two C1's. These reactions also give A1. A Norrish I reaction on K1 can also occur, which leads to the formation of carbon dioxide and R3. As shown in the case of polypropylene,33isomerization to a more stable tertiary radical occurs on R3. The oxidation of this radical following a classical mechanism is the route to produce acetic acid, which regenerates the primary alkyl radical R3. The recombination of two R3 radicals, followed by two successive oxidations on two carbon 1’s, is responsible for the formation of propanoic acid and regenerates R3 and A1. Hydrogen abstraction at carbon 1 on A1 leads to the formation of a macroradical, which can evolve in two different ways. A first β-scission (1) provokes the formation of malonic acid and R3. The second β-scission (2) regenerates A1 and forms a radical, which evolves by recombination to form succinic acid or by hydrogen abstraction to form acetic acid. It should be noted that β-scission on a C–O bond can also occur to form the chain ketone K1. This path is not represented in Scheme 1. Hydrogen abstraction at C2 on A1 can also occur. By the same β-scissions previously described (3 and 4), oxalic acid and formic acid are formed. Even if less favorable, radical attack on the secondary carbon atom (carbon 2) can also occur. The following mechanism accounting for the oxidation on the secondary carbon (C2) of PVA can be proposed (Scheme 2).
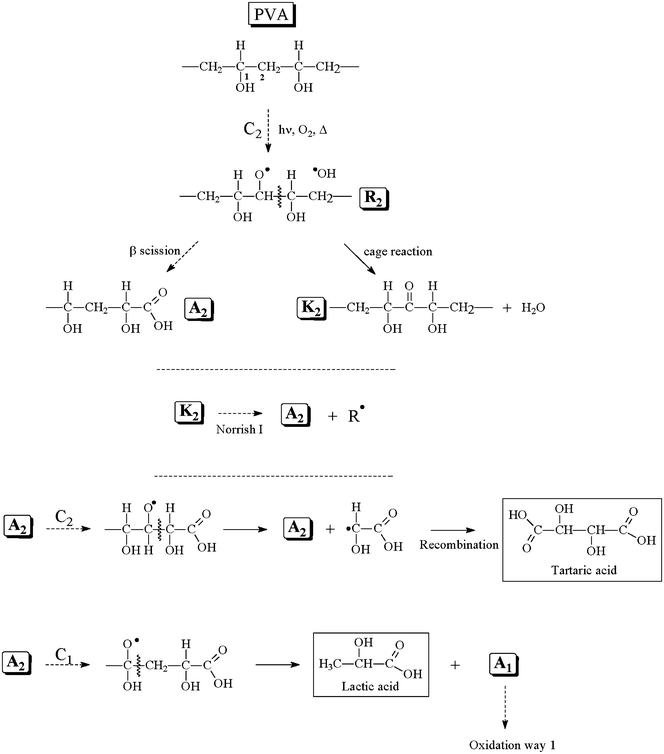 | ||
| Scheme 2 Oxidation mechanism of the PVA with a primary attack on the secondary carbon (C2). | ||
A hydrogen abstraction at C2 leads to the formation of a macroalkyl radical, which forms hydroperoxides as primary oxidation products. The photochemical decomposition of the hydroperoxides leads to alkoxy radicals (R2). β-Scission on R2 occurs to form an aldehyde, which can be oxidized to an α-hydroxylated carboxylic acid (A2). A cage reaction on R2 also occurs to form a chain ketone (K2) and water. K2 and A2 are formed as the secondary products in this way. Norrish I reactions on K2 can form carbon dioxide and regenerate A2. As previously observed for A1, A2 can evolve in two different ways. The first implies hydrogen abstraction on carbon 1 followed by chain scission to regenerate A2 and form a radical, which creates tartaric acid by recombination. Hydrogen abstraction on C1 can also occur and is responsible for the formation of lactic acid. This last reaction also produces A1, which can be oxidized following Scheme 1. These two schemes show that A1, A2 and R3 are always regenerated during photooxidation (A2 can also produce A1). The propagation of these reactions leads to the formation of notable concentrations of low molecular weight products, which can migrate from the solid polymeric matrix. R3 is mainly responsible for the formation of acetic acid, whereas A1 principally causes the formation of oxalic, malonic and succinic acids. These products are the major products detected in ionic chromatography.
4. Conclusion
In this paper, a comprehensive study of PVA photooxidation led to a proposal for the mechanism to account for the modifications of the chemical structure of PVA upon irradiation. The obtained results indicate that oxidized products are formed in the first 5 microns at the surface of the exposed films and are mainly carboxylic acids. Furthermore, numerous low molecular weight products were identified that can be trapped in the film or migrate in the gas phase. This could provoke a progressive erosion of the surface. The strongest point is the high stability of PVA upon irradiation in the absence of oxygen, even after long exposure in conditions of accelerated ageing. This result is of prime importance for the incorporation of PVA in a durable multilayer encapsulation system for OSCs. Therefore, PVA appears to be a good organic candidate for OSC encapsulation with high durability, as long as the PVA layer is protected from air by an inorganic layer as a first outside layer in the inorganic/organic multistack.References
- R. Jayasekara, I. Harding, I. Bowater, G. B. Y. Christie and G. T. Lonergan, Polym. Test., 2004, 23, 17–27 CrossRef CAS.
- M. Kokabi, M. Sirousazar and Z. M. Hassan, Eur. Polym. J., 2007, 43, 773–781 CrossRef CAS.
- H. S. Mansur, C. M. Sadahira, A. N. Souza and A. A. P. Mansur, Mater. Sci. Eng., C, 2008, 28, 539–548 CrossRef CAS.
- C. Charton, N. Schiller, M. Fahland, A. Holländer, A. Wedel and K. Noller, Thin Solid Films, 2006, 502, 99–103 CrossRef CAS.
- J. Brandrup, E. H. Immergut and E. A. Grulke, Polymer Handbook, 1999 Search PubMed.
- G. Dennler, C. Lungenschmied, H. Neugebauer, N. S. Sariciftci, M. Latrèche, G. Czeremuszkin and M. R. Wertheimer, Thin Solid Films, 2006, 511-512, 349–353 CrossRef.
- M. S. Weaver, L. A. Michalski, K. Rajan, M. A. Rothman, J. A. Silvernail, J. J. Brown, P. E. Burrows, G. L. Graff, M. E. Gross, P. M. Martin, M. Hall, E. Mast, C. Bonham, W. Bennett and M. Zumhoff, Appl. Phys. Lett., 2002, 81, 2929–2931 CrossRef CAS.
- D. S. Wuu, T. N. Chen, C. C. Wu, C. C. Chiang, Y. P. Chen, R. H. Horng and F. S. Juang, Chem. Vap. Deposition, 2006, 12, 220–224 CrossRef CAS.
- T. N. Chen, D. S. Wuu, C. C. Wu, C. C. Chiang, Y. P. Chen and R. H. Horng, Plasma Processes Polym., 2007, 4, 180–185 Search PubMed.
- A. Sionkowska, J. Skopinska and M. Wisniewski, Polym. Degrad. Stab., 2004, 83, 117–125 CrossRef CAS.
- A. Sionkowska, A. Planecka, J. Kozlowska and J. Skopinska-Wisniewska, Polym. Degrad. Stab., 2009, 94, 383–388 CrossRef CAS.
- H. Kaczmarek and A. Podgórski, J. Photochem. Photobiol., A, 2007, 191, 209–215 CrossRef CAS.
- H. Kaczmarek and A. Podgórski, Polym. Degrad. Stab., 2007, 92, 939–946 CrossRef CAS.
- f. Djouani, Y. Israëli, L. Frezet, A. Rivaton, R. A. Lessard and M. Bolte, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1317–1325 CrossRef CAS.
- J. Lemaire, R. Arnaud and J.-L. Gardette, Rev. Gen. Caout. Plast., 1981, 613, 87–92 Search PubMed.
- D. J. Carlsson, R. Brousseau, C. Zhang, D. M. Wiles, in ACS Symposium Series, American Chemical Society, Anheim, 1988, pp. 376–389 Search PubMed.
- C. Wilhelm J.-L. Gardette, Wiley Subscription Services, Inc., A Wiley Company, 1994, pp. 1411–1420 Search PubMed.
- B. J. Holland and J. N. Hay, Polymer, 2001, 42, 6775–6783 CrossRef CAS.
- K. Maruyama, H. Akahoshi, M. Kobayashi and Y. Tanizaki, Bull. Chem. Soc. Jpn., 1985, 58, 2923–2928 CrossRef CAS.
- K. Maruyama, H. Akahoshi, M. Kobayashi and Y. Tanizaki, Chem. Lett., 1983, 12, 1863–1866 CrossRef.
- J. L. Gardette, S. Gaumet and J. Lemaire, Macromolecules, 1989, 22, 2576–2581 CrossRef CAS.
- S. Gaumet and J. L. Gardette, Polym. Degrad. Stab., 1991, 33, 17–34 CrossRef CAS.
- P. R. Somani, R. Marimuthu, A. K. Viswanath and S. Radhakrishnan, Polym. Degrad. Stab., 2003, 79, 77–83 CrossRef CAS.
- N. V. Bhat, M. M. Nate, M. B. Kurup, V. A. Bambole and S. Sabharwal, Nucl. Instrum Meth. B, 2005, 237, 585–592 CrossRef CAS.
- G. Socrates, Infrared and Raman Characteristic Group Frequencies, 2001 Search PubMed.
- A. Roger, D. Sallet and J. Lemaire, Macromolecules, 1985, 18, 1771–1775 CrossRef CAS.
- A. Rivaton, Polym. Degrad. Stab., 1993, 41, 297–310 CrossRef CAS.
- S. Akhter, K. Allan, D. Buchanan, J. A. Cook, A. Campion and J. M. White, Appl. Surf. Sci., 1988, 35, 241–258 CrossRef CAS.
- J.-L. Gardette, in Handbook Polym. Degrad., ed. S. Halim Hamid, Marcel Dekker, Inc., New York, 2000, pp. 671–698 Search PubMed.
- D. J. Carlsson and D. M. Wiles, Macromolecules, 1969, 2, 587–597 CrossRef CAS.
- J.-L. Philippart, F. Posada and J.-L. Gardette, Polym. Degrad. Stab., 1995, 49, 285–290 CrossRef CAS.
- J.-L. Philippart, F. Posada and J.-L. Gardette, Polym. Degrad. Stab., 1996, 53, 33–37 CrossRef CAS.
- P. Delprat, X. Duteurtre and J.-L. Gardette, Polym. Degrad. Stab., 1995, 50, 1–12 CrossRef CAS.
Footnote |
| † Current address: Laboratoire de Photochimie Moléculaire et Macromoléculaire, UMR CNRS/Université Blaise Pascal 6505, 24 avenue des Landais, BP 80026, 63171 Aubière Cedex, France. |
| This journal is © The Royal Society of Chemistry 2011 |