Microwave-assisted one-pot synthesis of 2-aryl-5,6-dihydro-4H-1,3-thiazines via reaction between Lawesson’s reagent and allyl arylamides derived from Morita–Baylis–Hillman acetates†‡
Subhendu
Bhowmik§
,
Amita
Mishra§
and
Sanjay
Batra
*
Medicinal and Process Chemistry Division, CSIR-Central Drug Research Institute (CSIR), PO Box 173, Lucknow, 226001, India. E-mail: batra_san@yahoo.co.uk; s_batra@cdri.res.in; Fax: +91-522-2623405; Tel: +91-522-2621411-18 Extn. 4234, 4368
First published on 16th September 2011
Abstract
A one-pot synthesis of 2-aryl-5,6-dihydro-4H-1,3-thiazines from the allyl arylamides afforded from the Morita–Baylis–Hillman acetates of acrylates has been developed. The protocol comprises of Lawesson’s reagent-mediated transformation of allyl arylamide to thioamide followed by tandem intramolecular sulfa-Michael reaction under microwave condition to afford the products as a mixture of syn and anti isomers. Based on a plausible mechanism syn and anti-isomers are proposed as the kinetic and thermodynamic products, respectively. Further it has been experimentally demonstrated that the syn isomer is transformed to the anti isomer via prolonged heating.
Introduction
The proclivity of the Morita–Baylis–Hillman (MBH) reaction to yield densely functionalized products in atom-economical fashion has made it extremely popular with respect to synthetic organic chemistry. Several derivatives, which are readily prepared from the MBH adducts, serve as starting materials in diverse complexity generating reactions leading to an array of molecular frameworks.1 In particular they have been demonstrated to be versatile precursors to a variety of aza- and oxa-heterocycles. Intriguingly however, there are only a few reports related to synthesis of sulfa-heterocycles employing the MBH derivatives.2 In our research program related to the development of cyclic compounds from the MBH derivatives we have successfully developed a facile one-pot diastereoselective synthesis of 2-substituted amino-5,6-dihydro-1,3-thiazines from the primary allylamines afforded by the MBH acetates.3 The two step protocol proceed via initial reaction between the primary allylamines and aryl isothiocyanates to produce allyl thiourea followed by intramolecular sulfa-Michael reaction.5,6-Dihydro-1,3-thiazines represent core structure of several bioactive compounds, therefore development of simple and general approach to this structural motif is attractive target for chemists.4 The success of our method to obtain 2-substituted amino-5,6-dihydro-1,3-thiazines coupled with our interest to enhance the scope of MBH chemistry in realms of heterocyclic synthesis, led us to envisage the synthesis of 2-aryl-5,6-dihydro-1,3-thiazines from the primary allylmines. Our reasoning is based on the retrosynthetic plan outlined in Scheme 1. An aroylation reaction with the primary allylamine should give the allyl arylamide which on reaction with Lawesson’s reagent (LR)5 may produce allyl arylthioamide which would undergo intramolecular cyclizationvia a sulfa-Michael reaction. During the course of our study towards this objective we have observed that the allyl arylamides afforded from the MBH derivative of acrylates successfully affords the desired 1,3-thiazines whereas the allyl arylamides derived from the MBH derivative of acrylonitrile produced a complex mixture of products. Herein, we wish to present the details of the results of this study.
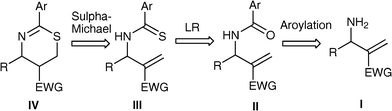 | ||
| Scheme 1 Retrosynthetic plan for the synthesis of aryl-5,6-dihydro-1,3-thiazine from allyl arylamide. | ||
Results and Discussion
The allylamines 3a–h and 4a–e were prepared following the reported procedure. Treating the allylamines with benzoyl chloride furnished the corresponding allylbenzamides 5a–h and 6a–e in good yields (Scheme 2). Initial experiments for the purpose of optimization were performed using 5d as the model substrate. Guided by our experience we anticipated that conversion of benzamide to benzthioamideviaLR could either lead to spontaneous intramolecular sulfa-Michael reaction or may require a Lewis-acid as additive to afford the desired 1,3-thiazine. Accordingly the reaction of 5d with LR was investigated by employing different conditions with respect to solvents, temperature and duration. We were pleased to observe that the reaction was successful in diphenyl ether as medium under heating at 150 °C for 2 h to afford a mixture of two products. Subsequent isolation viasilica gel chromatography and characterization revealed that the higher Rf compound obtained was the anti-isomer of expected 1,3-thiazine, 7d, whereas the lower Rf compound was the syn-isomer of 7d, in a 30% combined yield. The syn:anti ratio was found to be 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The relative stereochemistry of 7d was established on the basis of coupling constant of the CH-protons at C-5 and C-6. The J value of the CH proton in anti-7d was 12.1 Hz whereas in syn-7d it was observed to be 4.7 Hz. The stereochemistry of the two isomers was further corroborated viaNOESY experiments. Although we succeeded in synthesizing the desired product in one-pot, low isolated yields warranted further examining of the reaction conditions. A series of reactions investigating the use of different solvents and temperature under microwave irradiation was thus conducted. To our delight, the reaction of 5d with LR in diphenyl ether under microwave irradiation (MW) at 150 °C was complete in 8 min and the combined isolated yields of syn and anti isomers of 7d increased from 30 to 57% (syn:anti, 2:1). The reaction in toluene though took 30 min to go to completion at 110 °C, the yields increased to 60% (syn:anti, 2:1) whereas the reaction in xylene at 150 °C was complete in 8 min and the isolated yields of 7d enhanced to 65% (syn:anti, 2:1). We experienced that as compared to diphenyl ether, the work up of reaction performed in xylene as medium was convenient and straightforward.
:1. The relative stereochemistry of 7d was established on the basis of coupling constant of the CH-protons at C-5 and C-6. The J value of the CH proton in anti-7d was 12.1 Hz whereas in syn-7d it was observed to be 4.7 Hz. The stereochemistry of the two isomers was further corroborated viaNOESY experiments. Although we succeeded in synthesizing the desired product in one-pot, low isolated yields warranted further examining of the reaction conditions. A series of reactions investigating the use of different solvents and temperature under microwave irradiation was thus conducted. To our delight, the reaction of 5d with LR in diphenyl ether under microwave irradiation (MW) at 150 °C was complete in 8 min and the combined isolated yields of syn and anti isomers of 7d increased from 30 to 57% (syn:anti, 2:1). The reaction in toluene though took 30 min to go to completion at 110 °C, the yields increased to 60% (syn:anti, 2:1) whereas the reaction in xylene at 150 °C was complete in 8 min and the isolated yields of 7d enhanced to 65% (syn:anti, 2:1). We experienced that as compared to diphenyl ether, the work up of reaction performed in xylene as medium was convenient and straightforward.
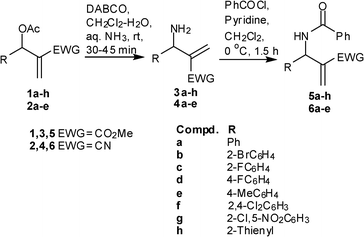 | ||
| Scheme 2 Synthesis of allyl benzamides. | ||
Having successfully optimized the conditions, scope of the methodology was tested by reacting other allylbenzamides 5a–c,e–h and 6a–e with LR under MW irradiation. It was observed that amides belonging to series 5 furnished the respective 1,3-thiazines 7 whereas amides belonging to series 6 produced a mixture of products which were difficult to isolate.6 As delineated in Table 1, amides 5a–c,e–h gave the corresponding 1,3-thiazines 7a–c,e–h as a mixture of syn:anti isomers in an approximate ratio of 2:1. Except for 7f–g (entry 6–7), the mixture of syn and anti isomers were easily separated viasilica gel column chromatography. Against the observed trend, the amide 5b furnished the anti-isomer of 7b only (entry 2).
|
|
|||
|---|---|---|---|
| Entry | R | syn 7 (yield%) a | anti 7 (yield%) a |
| a Isolated yields after column chromatography. | |||
| 1 | Ph | 47 | 21 |
| 2 | 2-BrC6H4 | – | 63 |
| 3 | 2-FC6H4 | 40 | 22 |
| 4 | 4-FC6H4 | 41 | 24 |
| 5 | 4-MeC6H4 | 40 | 23 |
| 6 | 2,4-Cl2C6H4 | 68 (inseparable) | 68 (inseparable) |
| 7 | 2-Cl2,4-NO2C6H4 | 72 (inseparable) | 72 (inseparable) |
| 8 | 2-Thienyl | 41 | 29 |
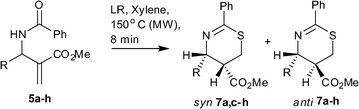
In order to address the stereochemical outcome of the reaction we speculate that perhaps the possible mechanism for this series is similar to the that of aminothiazines reported earlier. The initial nucleophilic attack of sulfur of the thioamide results into an enolate intermediate as shown in Scheme 3. Protonation of the enolate intermediate from the less hindered side leads to kinetically controlled syn-isomer. Because the reaction under MW is complete in a short period we presume that the results in Table 1 reflect the kinetic ratios. Nevertheless to seek experimental support for this hypothetical assumption we carried out reactions under controlled conditions via conventional heating. Accordingly reactions of compounds 5b and 5d with LR in xylene at 140 °C were examined at different time intervals. The presence of only the kinetic product i.e. the syn-isomer along with the starting material was detected after 30 min of reaction time for these reactions. The reaction for 5b was complete in 2 h to afford the product as mixture of syn-7b and anti-7b in 1:5 ratio (caTable 1, entry 2). However, pursuing the reaction till 24 h resulted in anti-7b, exclusively. From the comparison drawn between results of conventional and MW heating for 5b, it is assumed that under MW the presence of a bromo substitution at 2-position of the phenyl ring destabilizes the kinetic-controlled syn-product which undergoes fast inversion to the thermodynamically stable anti-product. In contrast to 5b, the reaction of 5d was complete after 2.5 h to furnish the product as a mixture of syn-7d and anti-7d in 2:1 ratio. Nevertheless here too prolonging the reaction to 36 h, altered the ratio of syn and anti isomer to 1:5 (caTable 1, entry 4). Hence these results imply that the anti-isomer is the thermodynamic product and is more stable form of these 1,3-thiazines. In order to evaluate whether the syn-isomer can be transformed to the anti-isomer experimentally, in a representative study syn-7d was heated in xylene with and without LR. After 36 h the syn-7d was transformed to anti-7d under the influence of LR whereas simple heating in the absence of LR did not induce any change and the starting material was recovered.
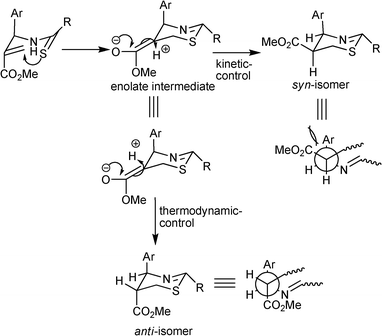 | ||
| Scheme 3 Plausible mechanism explaining the formation of a mixture of syn and anti isomers of 1,3-thiazines from allyl arylthioamide. | ||
Having examined the scope of our methodology with allyl benzamides, it was considered essential to test the outcome with other arylamides prepared from different acid chlorides. Hence allyl arylamides 8A–E were prepared by reacting 3a with randomly chosen acid chlorides A–E and treated with LR in xylene under MW irradiation. It was observed that this variation had significant impact on the outcome of the reaction (Table 2). As observed with benzamide, the amides 8A–B,D afforded a mixture of syn- and anti-isomers of corresponding 1,3-thiazines 9A–B, D (entry 1,2 and 4). For 8B, in addition to syn- and anti-9B, another product was isolated in minor quantity (5%) which was spectrally characterized to be the allyl thioamide 10B (entry 2). The mixture of syn- and anti-mixture of 9D could not be separated in pure form (entry 4), whereas the reaction of 8C was diastereoselective to furnish the anti-9C exclusively in 56% yields (entry 3). Surprisingly however, the amide 8E afforded the thioamide 10E exclusively in 77% yields (entry 5). In our attempts to isolate the syn-9C, the arylamide 8C was subjected to reaction with LR under conventional heating. Unfortunately, however even after 72 h of reaction time we did not observe the formation of any product. To gain insight into the sequence of formation of thioamides 10, the amide 8E was heated under MW in the absence of LR. It was discovered that even after 30 min the starting material remained unchanged (Scheme 4). This implies that the 1,3-migration occurs after the formation of thioamideV. However, by arresting the reaction at different time intervals we were unable to isolate the intermediate V.
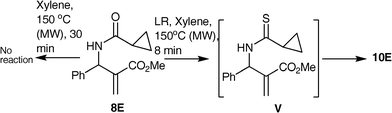 | ||
| Scheme 4 Results of the reactions of the thioamide 8E with and without LR. | ||
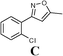
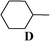
Conclusions
In summary, we have developed a new methodology to access 2-aryl-5,6-dihydro-1,3-thiazines. The route relies on sequential LR-mediated thioamide formation and intramolecular sulfa-Michael reaction starting from allyl arylamide. The required allyl arylamides can be readily prepared from the MBH acetates of acrylates. In general, though the products are produced as a mixture of syn- and anti-isomers they can be separated viacolumn chromatography. Further we demonstrated that the syn-isomer can be transformed to the more stable anti-isomer via prolonged heating. This work signifies the usefulness of the allylamines derived from MBH acetates for the synthesis of sulfur-containing heterocycles.Experimental
General Experimental
Melting points are uncorrected and were determined in capillary tubes on a Precision melting point apparatus containing silicon oil. IR spectra were recorded using a Perkin Elmer’s RX I FTIR spectrophotometer. 1H NMR and 13C NMR spectra were recorded either on a Bruker DPX-200 FT or Bruker Avance DRX-300 spectrometer, using TMS as an internal standard (chemical shifts in δ). The ESMS were recorded on MICROMASS Quadro-II LCMS system. The HRMS spectra were recorded as EI-HRMS on a JEOL system or as DART-HRMS (recorded as ES+) on a JEOL-AccuTOF JMS-T100 LC Mass spectrometer having DART (Direct Analysis in Real Time) source using 35Cl and 79Br for the mass calculation. Elemental analyses were performed on a Carlo Erba’s 108 or an Elementar’s Vario EL III microanalyzer. Microwave-mediated reactions were performed in Biotage initiator 2.5 microwave system. The room temperature varied between 21 °C and 35 °C. All solvents and reagents were used as supplied by the vendors without any further purification. All column chromatography were performed on silica gel 100–200 mesh otherwise stated. The yields for allylamides were calculated based on the acetate as the starting substrate.Specific experimental
:9) furnished the pure product 5a as a white solid (860 mg, 68%).
:30, v/v); νmax (KBr) 1658 (CONH), 1734 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 3.73 (s, 3H, OCH3), 6.03 (s, 1H, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 6.23 (d, 1H, J = 8.9 Hz, CH), 6.41 (s, 1H, CH2), 7.26–7.35 (m, 5H, ArH), 7.43–7.53 (m, 4H, ArH), 7.83–7.86 (m, 2H, ArH and NH); 13C NMR (50 MHz, CDCl3) δ = 52.2, 55.5, 119.1, 123.4, 126.6, 127.3, 127.8, 128.0, 128.8, 128.9, 129.9, 131.9, 134.4, 139.2, 139.8, 166.5, 166.7; mass (ES+) m/z = 296.1(M++1). Anal. Calcd. for C18H17NO3 (Exact mass: 295.1208); C, 73.20; H, 5.80; N, 4.74; Found: C, 73.01; H, 5.63; N, 4.86.
:9.5) afforded pure syn-7a (100 mg, 47%) and anti-7a (45 mg, 21%) as brown oils.
:20, v/v); νmax (Neat) 1731 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 3.07–3.25 (m, 3H, CH and CH2), 3.68 (s, 3H, OCH3), 5.72 (d, 1H, J = 3.7 Hz, CH), 7.17 (t, 1H, J = 1.8 Hz, ArH), 7.26–7.46 (m, 7H, ArH), 7.88 (d, 2H, J = 7.5 Hz, ArH); 13C NMR (50 MHz, CDCl3) δ = 30.0, 40.9, 52.2, 61.3, 126.8, 127.9, 128.2, 128.6, 131.0, 138.4, 139.2, 158.9, 171.9; mass (ES+) m/z = 312.2 (M++1); DART-HRMS Calcd. for C18H18NO2S 312.1058; Found: 312.1061.
:20, v/v); νmax (Neat) 1730 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 2.76–2.83 (m, 1H, CH), 3.15 (dd, 1H, J1 = 12.5 Hz, J2 = 3.7 Hz, CH2), 3.53 (dd, 1H, J1 = 12.6 Hz, J2 = 9.2 Hz, CH2), 3.57 (s, 3H, OCH3), 5.20 (d, 1H, J = 7.4 Hz, CH), 7.26–7.30 (m, 2H, ArH), 7.34–7.44 (m, 6H, ArH), 7.87 (d, 2H, J = 6.7 Hz, ArH); 13C NMR (75 MHz, CDCl3) δ = 26.5, 43.2, 52.1, 62.5, 126.7, 127.5, 127.6, 127.8, 128.1, 128.5, 128.7, 130.9, 142.0, 158.1, 171.9; mass (ES+) m/z = 312.2 (M++1); DART-HRMS Calcd. for C18H18NO2S 312.1058; Found: 312.1062.
:20, v/v); νmax (Neat) 1736 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 3.43–3.49 (m, 4H, CH and OCH3), 3.55–3.59 (m, 2H, CH2), 5.24 (d, 1H, J = 2.8 Hz, CH), 7.14–7.20 (m, 1H, ArH), 7.35–7.47 (m, 4H, ArH), 7.57 (d, 1H, J = 7.9 Hz, ArH), 7.71–7.74 (m, 1H, ArH), 7.90–7.92 (m, 2H, ArH); 13C NMR (75 MHz, CDCl3) δ 28.2, 38.6, 51.8, 61.6, 123.2, 125.7, 126.8, 127.4, 128.5, 129.1, 130.9, 131.3, 132.5, 138.9, 140.3, 160.9, 170.6; mass (ES+) m/z = 390.1 (M++1), 393.1 (M++2); DART-HRMS calcd for C18H17BrNO2S 390.0163; Found 390.0168.
:20, v/v); νmax (Neat) 1737 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 3.08–3.16 (m, 2H, CH and CH2), 3.41–3.49 (m, 1H, CH2), 3.69 (s, 3H, OCH3), 5.88 (d, 1H, J = 4.3 Hz, CH), 7.15 (t, 2H, J = 9.2 Hz, ArH), 7.31 (t, 1H, J = 7.3 Hz, ArH), 7.37–7.45 (m, 3H, ArH), 7.59 (t, 1H, J = 8.0 Hz, ArH), 7.86–7.92 (m, 2H, ArH); 13C NMR (50 MHz, CDCl3) δ = 29.8, 39.6, 52.6, 60.9, 123.1, 126.8, 127.8, 128.5, 129.2, 129.9, 131.0, 133.5, 138.9, 140.5, 159.1, 171.9; mass (ES+) m/z = 390.1 (M++1), 393.1 (M++3); DART-HRMS calcd for C18H17BrNO2S 390.0163; Found 390.0166.
:20, v/v); νmax (Neat) 1729 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 3.11–3.33 (m, 3H, CH and CH2), 3.65 (s, 3H, OCH3), 5.78 (d, 1H, J = 3.2 Hz, CH), 7.02–7.16 (m, 2H, ArH), 7.24–7.34 (m, 2H, ArH), 7.38–7.46 (m, 3H, ArH), 7.87–7.90 (m, 2H, ArH); 13C NMR (75 MHz, CDCl3) δ = 29.8, 42.2, 52.5, 58.2, 114.9 (J = 17.1 Hz), 122.4 (J = 17.5 Hz), 125.8, 126.4, 127.2, 128.2, 128.9, 129.7, 129.8, 130.0, 138.1, 157.0, 157.7, 171.4; mass (ES+) m/z = 330.4 (M++1); DART-HRMS calcd for C18H17FNO2S 330.0964; Found 330.0965.
:20, v/v); νmax (Neat) 1728 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 2.96–3.02 (m, 1H, CH), 3.14 (dd, 1H, J1 = 12.6 Hz, J2 = 3.6 Hz, CH2), 3.49 (dd, 1H, J1 = 12.6 Hz, J2 = 8.5 Hz, CH2), 3.62 (s, 3H, OCH3), 5.59 (d, 1H, J = 6.7 Hz, CH), 7.04–7.24 (m, 3H, ArH), 7.25–7.32 (m, 1H, ArH), 7.36–7.44 (m, 3H, ArH), 7.83–7.90 (m, 2H, ArH); 13C NMR (75 MHz, CDCl3) δ = 29.8, 41.1, 52.4, 56.6, 115.7 (J = 32.4 Hz), 124.4(J = 5.1 Hz), 126.7, 126.8, 128.5, 129.3, 129.5, 129.7, 130.9, 138.9, 158.9, 162.7, 172.4; mass (ES+) m/z = 330.1 (M++1); DART-HRMS calcd for C18H17FNO2S 330.0964; Found 330.0967.
:20, v/v); νmax (Neat) 1734 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 3.13–3.16 (m, 3H, CH and CH2), 3.68 (s, 3H, OCH3), 5.69 (d, 1H, J = 4.7 Hz, CH), 6.97–7.18 (m, 4H, ArH), 7.40–7.47 (m, 3H, ArH), 7.85–7.89 (m, 2H, ArH); 13C NMR (75 MHz, CDCl3) δ = 22.4, 40.8, 52.2, 60.5, 115.3, 115.6, 126.7, 128.5, 128.6, 129.4, 129.6, 131.0, 134.2, 138.8, 159.1, 164.3, 171.7; mass (ES+) m/z = 330.1 (M++1); DART-HRMS calcd. for C18H18NO2S 330.0964; Found 330.0968.
:20, v/v); νmax (Neat) 1734 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 2.67–2.78 (m, 1H, CH), 3.14 (dd, 1H, J1 = 18.8 Hz, J2 = 5.6 Hz, CH2), 3.47–3.58 (m, 4H, CH2 and OCH3), 5.09 (d, 1H, J = 12.1 Hz, CH), 7.00–7.10 (m, 3H, ArH), 7.21–7.31 (m, 2H, ArH), 7.37–7.44 (m, 2H, ArH), 7.83–7.88 (m, 2H, ArH); 13C NMR (75 MHz, CDCl3) δ = 26.9, 43.5, 52.3, 62.1, 115.3, 115.6, 119.0, 123.3, 126.7, 129.2 (J = 8.1 Hz), 129.8, 131.0, 138.0 (J = 3.1 Hz), 138.7, 158.4, 163.9, 173.2; mass (ES+) m/z = 330.0 (M++1); DART-HRMS calcd. for C18H18NO2S 330.0964; Found 330.0969.
:20, v/v); νmax (Neat) 1729 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 2.32 (s, 3H, CH3), 2.36 (d, 1H, J = 9.9 Hz, CH), 3.04–3.18 (m, 2H, CH2), 3.69 (s, 3H, OCH3), 5.70 (d, 1H, J = 2.5 Hz, CH), 7.03 (d, 2H, J = 8.0 Hz, ArH), 7.13 (t, 2H, J = 6.7 Hz, ArH), 7.38–7.45 (m, 3H, ArH), 7.86–7.88 (m, 2H, ArH); 13C NMR (75 MHz, CDCl3) δ = 21.1, 29.7, 40.7, 51.9, 60.8, 126.5, 127.6, 128.3, 128.4, 129.1, 130.7, 135.1, 137.7, 154.2, 171.9; mass (ES+) m/z = 326.0 (M++1); DART-HRMS calcd. for C19H20NO2S 326.1215; Found 326.1218.
:20, v/v); νmax (Neat) 1735 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 2.34 (s, 3H, CH3), 2.75–2.82 (m, 1H, CH), 3.15 (dd, 1H, J1 = 12.5 Hz, J2 = 3.7 Hz, CH2), 3.48 (dd, 1H, J1 = 12.2 Hz, J2 = 8.7 Hz, CH2), 3.59 (s, 3H, OCH3), 5.19 (d, 1H, J = 7.3 Hz, CH), 7.15 (brs, 4H, ArH), 7.36–7.44 (m, 3H, ArH), 7.86 (d, 2H, J = 6.7 Hz, ArH); 13C NMR (75 MHz, CDCl3) δ = 21.1, 26.3, 43.1, 52.2, 62.0, 126.6, 127.2, 128.3, 129.2, 130.7, 137.1, 138.8, 138.9, 157.7, 173.0; mass (ES+) m/z = 326.1(M++1); DART-HRMS calcd. for C19H20NO2S 326.1215; Found 326.1220.
CH2), 6.23 (d, 1H, J = 8.9 Hz, CH), 6.41 (s, 1H, CH2), 7.26–7.35 (m, 5H, ArH), 7.43–7.53 (m, 4H, ArH), 7.83–7.86 (m, 2H, ArH and NH); 13C NMR (50 MHz, CDCl3) δ = 52.2, 55.5, 119.1, 123.4, 126.6, 127.3, 127.8, 128.0, 128.8, 128.9, 129.9, 131.9, 134.4, 139.2, 139.8, 166.5, 166.7; mass (ES+) m/z = 296.1(M++1). Anal. Calcd. for C18H17NO3 (Exact mass: 295.1208); C, 73.20; H, 5.80; N, 4.74; Found: C, 73.01; H, 5.63; N, 4.86.
:9.5) afforded pure syn-7a (100 mg, 47%) and anti-7a (45 mg, 21%) as brown oils.
:20, v/v); νmax (Neat) 1731 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 3.07–3.25 (m, 3H, CH and CH2), 3.68 (s, 3H, OCH3), 5.72 (d, 1H, J = 3.7 Hz, CH), 7.17 (t, 1H, J = 1.8 Hz, ArH), 7.26–7.46 (m, 7H, ArH), 7.88 (d, 2H, J = 7.5 Hz, ArH); 13C NMR (50 MHz, CDCl3) δ = 30.0, 40.9, 52.2, 61.3, 126.8, 127.9, 128.2, 128.6, 131.0, 138.4, 139.2, 158.9, 171.9; mass (ES+) m/z = 312.2 (M++1); DART-HRMS Calcd. for C18H18NO2S 312.1058; Found: 312.1061.
:20, v/v); νmax (Neat) 1730 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 2.76–2.83 (m, 1H, CH), 3.15 (dd, 1H, J1 = 12.5 Hz, J2 = 3.7 Hz, CH2), 3.53 (dd, 1H, J1 = 12.6 Hz, J2 = 9.2 Hz, CH2), 3.57 (s, 3H, OCH3), 5.20 (d, 1H, J = 7.4 Hz, CH), 7.26–7.30 (m, 2H, ArH), 7.34–7.44 (m, 6H, ArH), 7.87 (d, 2H, J = 6.7 Hz, ArH); 13C NMR (75 MHz, CDCl3) δ = 26.5, 43.2, 52.1, 62.5, 126.7, 127.5, 127.6, 127.8, 128.1, 128.5, 128.7, 130.9, 142.0, 158.1, 171.9; mass (ES+) m/z = 312.2 (M++1); DART-HRMS Calcd. for C18H18NO2S 312.1058; Found: 312.1062.
:20, v/v); νmax (Neat) 1736 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 3.43–3.49 (m, 4H, CH and OCH3), 3.55–3.59 (m, 2H, CH2), 5.24 (d, 1H, J = 2.8 Hz, CH), 7.14–7.20 (m, 1H, ArH), 7.35–7.47 (m, 4H, ArH), 7.57 (d, 1H, J = 7.9 Hz, ArH), 7.71–7.74 (m, 1H, ArH), 7.90–7.92 (m, 2H, ArH); 13C NMR (75 MHz, CDCl3) δ 28.2, 38.6, 51.8, 61.6, 123.2, 125.7, 126.8, 127.4, 128.5, 129.1, 130.9, 131.3, 132.5, 138.9, 140.3, 160.9, 170.6; mass (ES+) m/z = 390.1 (M++1), 393.1 (M++2); DART-HRMS calcd for C18H17BrNO2S 390.0163; Found 390.0168.
:20, v/v); νmax (Neat) 1737 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 3.08–3.16 (m, 2H, CH and CH2), 3.41–3.49 (m, 1H, CH2), 3.69 (s, 3H, OCH3), 5.88 (d, 1H, J = 4.3 Hz, CH), 7.15 (t, 2H, J = 9.2 Hz, ArH), 7.31 (t, 1H, J = 7.3 Hz, ArH), 7.37–7.45 (m, 3H, ArH), 7.59 (t, 1H, J = 8.0 Hz, ArH), 7.86–7.92 (m, 2H, ArH); 13C NMR (50 MHz, CDCl3) δ = 29.8, 39.6, 52.6, 60.9, 123.1, 126.8, 127.8, 128.5, 129.2, 129.9, 131.0, 133.5, 138.9, 140.5, 159.1, 171.9; mass (ES+) m/z = 390.1 (M++1), 393.1 (M++3); DART-HRMS calcd for C18H17BrNO2S 390.0163; Found 390.0166.
:20, v/v); νmax (Neat) 1729 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 3.11–3.33 (m, 3H, CH and CH2), 3.65 (s, 3H, OCH3), 5.78 (d, 1H, J = 3.2 Hz, CH), 7.02–7.16 (m, 2H, ArH), 7.24–7.34 (m, 2H, ArH), 7.38–7.46 (m, 3H, ArH), 7.87–7.90 (m, 2H, ArH); 13C NMR (75 MHz, CDCl3) δ = 29.8, 42.2, 52.5, 58.2, 114.9 (J = 17.1 Hz), 122.4 (J = 17.5 Hz), 125.8, 126.4, 127.2, 128.2, 128.9, 129.7, 129.8, 130.0, 138.1, 157.0, 157.7, 171.4; mass (ES+) m/z = 330.4 (M++1); DART-HRMS calcd for C18H17FNO2S 330.0964; Found 330.0965.
:20, v/v); νmax (Neat) 1728 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 2.96–3.02 (m, 1H, CH), 3.14 (dd, 1H, J1 = 12.6 Hz, J2 = 3.6 Hz, CH2), 3.49 (dd, 1H, J1 = 12.6 Hz, J2 = 8.5 Hz, CH2), 3.62 (s, 3H, OCH3), 5.59 (d, 1H, J = 6.7 Hz, CH), 7.04–7.24 (m, 3H, ArH), 7.25–7.32 (m, 1H, ArH), 7.36–7.44 (m, 3H, ArH), 7.83–7.90 (m, 2H, ArH); 13C NMR (75 MHz, CDCl3) δ = 29.8, 41.1, 52.4, 56.6, 115.7 (J = 32.4 Hz), 124.4(J = 5.1 Hz), 126.7, 126.8, 128.5, 129.3, 129.5, 129.7, 130.9, 138.9, 158.9, 162.7, 172.4; mass (ES+) m/z = 330.1 (M++1); DART-HRMS calcd for C18H17FNO2S 330.0964; Found 330.0967.
:20, v/v); νmax (Neat) 1734 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 3.13–3.16 (m, 3H, CH and CH2), 3.68 (s, 3H, OCH3), 5.69 (d, 1H, J = 4.7 Hz, CH), 6.97–7.18 (m, 4H, ArH), 7.40–7.47 (m, 3H, ArH), 7.85–7.89 (m, 2H, ArH); 13C NMR (75 MHz, CDCl3) δ = 22.4, 40.8, 52.2, 60.5, 115.3, 115.6, 126.7, 128.5, 128.6, 129.4, 129.6, 131.0, 134.2, 138.8, 159.1, 164.3, 171.7; mass (ES+) m/z = 330.1 (M++1); DART-HRMS calcd. for C18H18NO2S 330.0964; Found 330.0968.
:20, v/v); νmax (Neat) 1734 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 2.67–2.78 (m, 1H, CH), 3.14 (dd, 1H, J1 = 18.8 Hz, J2 = 5.6 Hz, CH2), 3.47–3.58 (m, 4H, CH2 and OCH3), 5.09 (d, 1H, J = 12.1 Hz, CH), 7.00–7.10 (m, 3H, ArH), 7.21–7.31 (m, 2H, ArH), 7.37–7.44 (m, 2H, ArH), 7.83–7.88 (m, 2H, ArH); 13C NMR (75 MHz, CDCl3) δ = 26.9, 43.5, 52.3, 62.1, 115.3, 115.6, 119.0, 123.3, 126.7, 129.2 (J = 8.1 Hz), 129.8, 131.0, 138.0 (J = 3.1 Hz), 138.7, 158.4, 163.9, 173.2; mass (ES+) m/z = 330.0 (M++1); DART-HRMS calcd. for C18H18NO2S 330.0964; Found 330.0969.
:20, v/v); νmax (Neat) 1729 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 2.32 (s, 3H, CH3), 2.36 (d, 1H, J = 9.9 Hz, CH), 3.04–3.18 (m, 2H, CH2), 3.69 (s, 3H, OCH3), 5.70 (d, 1H, J = 2.5 Hz, CH), 7.03 (d, 2H, J = 8.0 Hz, ArH), 7.13 (t, 2H, J = 6.7 Hz, ArH), 7.38–7.45 (m, 3H, ArH), 7.86–7.88 (m, 2H, ArH); 13C NMR (75 MHz, CDCl3) δ = 21.1, 29.7, 40.7, 51.9, 60.8, 126.5, 127.6, 128.3, 128.4, 129.1, 130.7, 135.1, 137.7, 154.2, 171.9; mass (ES+) m/z = 326.0 (M++1); DART-HRMS calcd. for C19H20NO2S 326.1215; Found 326.1218.
:20, v/v); νmax (Neat) 1735 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 2.34 (s, 3H, CH3), 2.75–2.82 (m, 1H, CH), 3.15 (dd, 1H, J1 = 12.5 Hz, J2 = 3.7 Hz, CH2), 3.48 (dd, 1H, J1 = 12.2 Hz, J2 = 8.7 Hz, CH2), 3.59 (s, 3H, OCH3), 5.19 (d, 1H, J = 7.3 Hz, CH), 7.15 (brs, 4H, ArH), 7.36–7.44 (m, 3H, ArH), 7.86 (d, 2H, J = 6.7 Hz, ArH); 13C NMR (75 MHz, CDCl3) δ = 21.1, 26.3, 43.1, 52.2, 62.0, 126.6, 127.2, 128.3, 129.2, 130.7, 137.1, 138.8, 138.9, 157.7, 173.0; mass (ES+) m/z = 326.1(M++1); DART-HRMS calcd. for C19H20NO2S 326.1215; Found 326.1220.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :0.4).
68% as brown oil (71 mg from 100 mg); Rf = 0. 33 (hexanes:EtOAc, 80:20, v/v); νmax (Neat) 1731 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 3.01–3.12, (m, 4H, CH and CH2), 3.41–3.50 (m, 2H, CH2), 3.52 (s, 3H, OCH3), 3.67 (s, 3H, OCH3), 5.26 (d, 1H, J = 2.9 Hz, CH), 5.77 (d, 1H, J = 5.6 Hz, CH), 7.10 (d, 1H, J = 8.4 Hz, ArH), 7.24–7.27 (m, 3H, ArH), 7.29–7.32 (m, 2H, ArH), 7.36–7.48 (m, 6H, ArH), 7.65 (d, 1H, J = 8.5 Hz, ArH), 7.83–7.90 (m, 3H, ArH); 13C NMR (50 MHz, CDCl3) δ = 25.3, 27.8, 38.6, 39.9, 52.0, 52.6, 58.6, 58.7, 126.7, 127.2, 127.5, 128.5, 129.0, 129.8, 130.7, 131.0, 131.1, 132.0, 133.1, 133.5, 133.8, 134.1, 137.3, 137.8, 138.7, 159.4, 170.5, 171.9; mass (ES+) m/z = 380.1 (M++1), 382.1 (M++3); DART-HRMS calcd. for C18H16Cl2NO2S 380.0279; Found 380.0285.
:0.4).
72% as brown oil (75 mg from 100 mg); Rf = 0.35 (hexanes:EtOAc, 80:20, v/v); νmax (Neat) 1731 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 2.99–3.05 (m, 1H, CH), 3.14 (dd, 1H, J1 = 12.8 Hz, J2 = 3.5 Hz, CH2), 3.50 (s, 4H, CH and OCH3), 3.52–3.62 (m, 3H, CH2), 3.65 (s, 3H, OCH3), 5.15 (d, 1H, J = 3.0 Hz, CH), 5.74 (d, 1H, J = 6.9 Hz, CH), 7.38–7.49 (m, 6H, ArH), 7.54–7.60 (m, 3H, ArH), 7.82–7.85 (m, 2H, ArH), 7.92–7.95 (m, 1H, ArH), 8.10–8.14 (m, 4H, ArH); 13C NMR (50 MHz, CDCl3) δ = 28.7, 29.7, 38.5, 40.6, 52.0, 52.6, 59.2, 59.4, 123.3, 123.6, 125.0, 126.7, 128.6, 130.0, 130.8, 131.2, 138.3, 138.8, 139.8, 141.5, 147.0, 160.5, 169.7, 171.6; mass (ES+) m/z = 391.0 (M++1), 393.1 (M++3); DART-HRMS calcd for C18H16ClN2O4S 391.0519; Found 391.0522.
:20, v/v); νmax (Neat) 1733 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 3.08–3.32 (m, 3H, CH and CH2), 3.76 (s, 3H, OCH3), 5.99 (d, 1H, J = 1.9 Hz, CH), 6.84 (d, 1H, J = 3.4 Hz, ArH), 6.97 (t, 1H, J = 3.6 Hz, ArH), 7.22–7.26 (m, 1H, ArH), 7.37–7.46 (m, 3H, ArH), 7.85–7.87 (m, 2H, ArH); 13C NMR (50 MHz, CDCl3) δ = 22.7, 40.9, 52.3, 57.0, 125.3, 125.7, 126.7, 126.8, 127.0, 128.5, 130.9, 138.9, 141.2, 171.4; mass (ES+) m/z = 318.1 (M++1); DART-HRMS calcd. for C16H16NO2S2 318.0622; Found 318.0624.
:20, v/v); νmax (Neat) 1734 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 2.85–2.91 (m, 1H, CH), 3.22 (dd, 1H, J1 = 12.6 Hz, J2 = 3.8 Hz, CH2), 3.50 (dd, 1H, J1 = 12.5 Hz, J2 = 9.2 Hz, CH2), 3.68 (s, 3H, OCH3), 5.51 (d, 1H, J = 7.6 Hz, CH), 6.92–6.99 (m, 2H, ArH), 7.26–7.27 (m, 1H, ArH), 7.37–7.45 (m, 3H, ArH), 7.86–7.89 (m, 2H, ArH); 13C NMR (50 MHz, CDCl3) δ = 26.6, 43.5, 52.5, 58.3, 124.3, 125.1, 126.8, 128.5, 131.0, 138.7, 146.2, 158.5, 172.9; mass (ES+) m/z = 318.2 (M++1); DART-HRMS calcd. for C16H16NO2S2 318.0622; Found 318.0623.
:20, v/v); νmax (Neat) 1730 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 3.07–3.18 (m, 3H, CH and CH2), 3.68 (s, 3H, OCH3), 5.71 (d, 1H, J = 2.1 Hz, CH), 6.92–6.95 (m, 1H, ArH), 7.12–7.15 (m, 1H, ArH), 7.23–7.34 (m, 5H, ArH), 7.50 (s, 1H, ArH); 13C NMR (75 MHz, CDCl3) δ = 29.8, 41.0, 52.1, 60.3, 111.8, 127.4, 127.8, 128.3, 128.6, 128.7, 128.9, 137.8, 144.6, 150.7, 171.5; mass (ES+) m/z = 302.1 (M++1); DART-HRMS calcd. for C16H16NO3S 302.0851; Found 302.0850.
:20, v/v); νmax (Neat) 1731 (CO2Me) cm−1; 1H NMR (200 MHz, CDCl3) δ = 2.75–2.88 (m, 1H, CH), 3.13 (dd, 1H, J1 = 18.8 Hz, J2 = 5.5 Hz, CH2), 3.45 (dd, 1H, J1 = 18.8 Hz, J2 = 12.9 Hz, CH2), 3.58 (s, 3H, OCH3), 5.24 (d, 1H, J = 6.8 Hz, CH), 6.45–6.48 (m, 1H, ArH), 6.93 (d, 1H, J = 3.4 Hz, ArH), 7.22–7.50 (m, 5H, ArH), 7.50 (d, 1H, J = 0.5 Hz, ArH); 13C NMR (75 MHz, CDCl3) δ = 25.6, 43.5, 52.3, 61.8, 111.3, 111.6, 127.5, 127.7, 127.9, 128.7, 141.8, 144.4, 149.0, 151.3, 172.7; mass (ES+) m/z = 302.1 (M++1); DART-HRMS calcd. for C16H16NO3S 302.0851; Found 302.0855.
:20, v/v); νmax (Neat) 1725 (CO2Me) cm−1; 1H NMR (200 MHz, CDCl3) δ = 2.33 (s, 3H, CH3), 3.02–3.07 (m, 1H, CH), 3.18–3.22 (m, 2H, CH2), 3.69 (s, 3H, CH3), 5.77 (d, 1H, J = 5.0 Hz, CH), 6.95 (s, 1H, ArH), 7.09 (brs, 4H, ArH), 7.17–7.22 (m, 2H, ArH), 7.30 (d, 2H, J = 2.0 Hz, ArH), 7.35 (brs, 6H, ArH); 13C NMR (50 MHz, CDCl3) δ = 21.5, 29.8, 41.2, 52.0, 60.8, 105.7, 125.6, 127.1, 127.9, 128.2, 128.5, 128.7, 129.0, 129.3, 138.1, 138.6, 140.0, 144.7, 151.4, 154.1, 171.9; mass (ES+) m/z = 468.2 (M++1); DART-HRMS calcd. for C28H26N3O2S 468.1746; Found 468.1750.
:20, v/v); νmax (Neat) 1725 (CO2Me) cm−1; 1H NMR (200 MHz, CDCl3) δ = 2.32 (s, 3H, CH3), 2.81–2.91 (m, 1H, CH), 3.14 (dd, 1H, J1 = 12.6 Hz, J2 = 3.6 Hz, CH2), 3.47 (dd, 1H, J1 = 18.8 Hz, J2 = 13.2 Hz, CH2), 3.59 (s, 3H, CH3), 5.27 (d, 1H, J = 7.0 Hz, CH), 6.96 (s, 1H, ArH), 7.08 (brs, 5H, ArH), 7.30 (brs, 2H, ArH), 7.34 (brs, 7H, ArH); 13C NMR (75 MHz, CDCl3) δ = 21.4, 25.7, 43.6, 52.3, 61.8, 105.8, 125.6, 127.2, 127.5, 127.6, 127.7, 127.9, 128.7, 129.0, 129.3, 138.6, 140.1, 142.0, 144.7, 173.0; mass (ES+) m/z = 468.2 (M++1); DART-HRMS calcd. for C28H26N3O2S 468.1746; Found 468.1747.
:20, v/v); νmax (Neat) 1727 (CO2Me) cm−1; 1H NMR (200 MHz, CDCl3) δ = 2.86–2.93 (m, 1H, CH), 3.20 (dd, 1H, J1 = 12.7 Hz, J2 = 3.6 Hz, CH2), 3.46–3.57 (m, 1H, CH2), 3.62 (s, 3H, OCH3), 5.34 (d, 1H, J = 6.5 Hz, CH), 7.21–7.23 (m, 1H, ArH), 7.25 (s, 1H, ArH), 7.30–7.42 (m, 6H, ArH), 7.47–7.52 (m, 1H, ArH), 7.72–7.77 (m, 1H, ArH); 13C NMR (50 MHz, CDCl3) δ = 25.4, 52.2, 52.5, 61.8, 104.6, 127.1, 127.2, 127.3, 127.7, 127.9, 128.4, 128.8, 130.5, 131.1, 131.2, 133.0, 137.3, 140.8, 161.5, 172.2; mass (ES+) m/z = 413.1 (M++1); DART-HRMS calcd. for C21H18ClN2O3S 413.0727; Found 413.0731.
:1).
69% as colorless oil (73 mg from 100 mg); Rf = 0.32 (hexanes:EtOAc, 80:20, v/v); νmax (Neat) 1735 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 1.30–1.36 (m, 3H, CH2), 1.46–1.58 (m, 4H, CH2), 1.62–1.68 (m, 3H, CH2), 1.79–2.06 (m, 10H,CH2 and CH), 2.27–2.38 (m, 2H, CH), 2.61–2.67 (m, 1H, CH2), 2.89–3.07 (m, 4H, CH and CH2), 3.32 (dd, 1H, J1 = 12.5 Hz, J2 = 8.8 Hz, CH2), 3.54 (s, 3H, CH3), 3.65 (s, 3H, CH3), 4.96 (d, 1H, J = 7.2 Hz, CH), 5.47 (d, 1H, J = 3.3 Hz, CH), 7.06–7.09 (m, 2H, ArH), 7.16–7.19 (m, 2H, ArH), 7.22–7.35 (m, 6H, ArH); 13C NMR (75 MHz, CDCl3) δ = 22.0, 22.8, 25.9, 26.2, 29.5, 29.8, 31.3, 31.4, 31.5, 31.6, 41.0, 43.4, 50.3, 50.6, 51.9, 52.1, 60.3, 61.3, 127.3, 127.5, 127.7, 128.0, 128.4, 128.6, 138.4, 142.0, 166.4, 167.0, 172.0, 173.1; mass (ES+) m/z = 318.1 (M++1); DART-HRMS calcd. for C18H24NO2S 318.1528; Found 318.1531.
:20, v/v); νmax (Neat) 1711 (CO2Me) cm−1; 1H NMR (200 MHz, CDCl3) δ = 2.34 (s, 3H, CH3), 3.87 (s, 3H, OCH3), 4.90 (d, 2H, J = 5.0 Hz, CH2), 7.10 (brs, 3H, ArH), 7.25 (s, 1H, ArH), 7.28–7.31 (m, 3H, ArH), 7.34–7.38 (m, 5H, ArH), 7.41–7.43 (m, 3H, ArH), 7.98 (s, 1H, CH), 9.04 (s, 1H, NH); 13C NMR (75 MHz, CDCl3) δ = 21.4, 42.8, 52.5, 110.6, 125.5, 126.4, 126.8, 128.1, 128.2, 128.6, 128.7, 128.8, 128.9, 129.1, 129.3, 129.7, 129.9, 134.1, 138.7, 139.7, 140.1, 144.8, 145.0, 151.1, 151.2, 168.0, 187.3; mass (ES+) m/z = 468.1 (M++1); DART-HRMS calcd. for C28H26N3O2S 468.1746; Found 468.1747.
:20, v/v); νmax (Neat) 1715 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 0.87–0.94 (m, 2H, CH2), 1.28–1.30 (m, 2H, CH2), 1.74–1.83 (m, 1H, CH), 3.88 (s, 3H, OCH3), 4.81 (d, 2H, J = 5.0 Hz, CH2), 7.30 (t, 1H, J = 3.9 Hz, ArH), 7.39–7.41 (m, 4H, ArH), 7.83 (brs, 1H, NH), 7.91 (s, 1H, CH); 13C NMR (50 MHz, CDCl3) δ = 12.1, 24.6, 43.5, 52.6, 126.2, 127.3, 128.1, 128.9, 129.8, 129.9, 133.9, 144.4, 168.4, 206.6; mass (ES+) m/z = 276.2 (M++1); DART-HRMS calcd. for C15H18NO2S 276.1058; Found 276.1060.
:0.4).
68% as brown oil (71 mg from 100 mg); Rf = 0. 33 (hexanes:EtOAc, 80:20, v/v); νmax (Neat) 1731 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 3.01–3.12, (m, 4H, CH and CH2), 3.41–3.50 (m, 2H, CH2), 3.52 (s, 3H, OCH3), 3.67 (s, 3H, OCH3), 5.26 (d, 1H, J = 2.9 Hz, CH), 5.77 (d, 1H, J = 5.6 Hz, CH), 7.10 (d, 1H, J = 8.4 Hz, ArH), 7.24–7.27 (m, 3H, ArH), 7.29–7.32 (m, 2H, ArH), 7.36–7.48 (m, 6H, ArH), 7.65 (d, 1H, J = 8.5 Hz, ArH), 7.83–7.90 (m, 3H, ArH); 13C NMR (50 MHz, CDCl3) δ = 25.3, 27.8, 38.6, 39.9, 52.0, 52.6, 58.6, 58.7, 126.7, 127.2, 127.5, 128.5, 129.0, 129.8, 130.7, 131.0, 131.1, 132.0, 133.1, 133.5, 133.8, 134.1, 137.3, 137.8, 138.7, 159.4, 170.5, 171.9; mass (ES+) m/z = 380.1 (M++1), 382.1 (M++3); DART-HRMS calcd. for C18H16Cl2NO2S 380.0279; Found 380.0285.
:0.4).
72% as brown oil (75 mg from 100 mg); Rf = 0.35 (hexanes:EtOAc, 80:20, v/v); νmax (Neat) 1731 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 2.99–3.05 (m, 1H, CH), 3.14 (dd, 1H, J1 = 12.8 Hz, J2 = 3.5 Hz, CH2), 3.50 (s, 4H, CH and OCH3), 3.52–3.62 (m, 3H, CH2), 3.65 (s, 3H, OCH3), 5.15 (d, 1H, J = 3.0 Hz, CH), 5.74 (d, 1H, J = 6.9 Hz, CH), 7.38–7.49 (m, 6H, ArH), 7.54–7.60 (m, 3H, ArH), 7.82–7.85 (m, 2H, ArH), 7.92–7.95 (m, 1H, ArH), 8.10–8.14 (m, 4H, ArH); 13C NMR (50 MHz, CDCl3) δ = 28.7, 29.7, 38.5, 40.6, 52.0, 52.6, 59.2, 59.4, 123.3, 123.6, 125.0, 126.7, 128.6, 130.0, 130.8, 131.2, 138.3, 138.8, 139.8, 141.5, 147.0, 160.5, 169.7, 171.6; mass (ES+) m/z = 391.0 (M++1), 393.1 (M++3); DART-HRMS calcd for C18H16ClN2O4S 391.0519; Found 391.0522.
:20, v/v); νmax (Neat) 1733 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 3.08–3.32 (m, 3H, CH and CH2), 3.76 (s, 3H, OCH3), 5.99 (d, 1H, J = 1.9 Hz, CH), 6.84 (d, 1H, J = 3.4 Hz, ArH), 6.97 (t, 1H, J = 3.6 Hz, ArH), 7.22–7.26 (m, 1H, ArH), 7.37–7.46 (m, 3H, ArH), 7.85–7.87 (m, 2H, ArH); 13C NMR (50 MHz, CDCl3) δ = 22.7, 40.9, 52.3, 57.0, 125.3, 125.7, 126.7, 126.8, 127.0, 128.5, 130.9, 138.9, 141.2, 171.4; mass (ES+) m/z = 318.1 (M++1); DART-HRMS calcd. for C16H16NO2S2 318.0622; Found 318.0624.
:20, v/v); νmax (Neat) 1734 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 2.85–2.91 (m, 1H, CH), 3.22 (dd, 1H, J1 = 12.6 Hz, J2 = 3.8 Hz, CH2), 3.50 (dd, 1H, J1 = 12.5 Hz, J2 = 9.2 Hz, CH2), 3.68 (s, 3H, OCH3), 5.51 (d, 1H, J = 7.6 Hz, CH), 6.92–6.99 (m, 2H, ArH), 7.26–7.27 (m, 1H, ArH), 7.37–7.45 (m, 3H, ArH), 7.86–7.89 (m, 2H, ArH); 13C NMR (50 MHz, CDCl3) δ = 26.6, 43.5, 52.5, 58.3, 124.3, 125.1, 126.8, 128.5, 131.0, 138.7, 146.2, 158.5, 172.9; mass (ES+) m/z = 318.2 (M++1); DART-HRMS calcd. for C16H16NO2S2 318.0622; Found 318.0623.
:20, v/v); νmax (Neat) 1730 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 3.07–3.18 (m, 3H, CH and CH2), 3.68 (s, 3H, OCH3), 5.71 (d, 1H, J = 2.1 Hz, CH), 6.92–6.95 (m, 1H, ArH), 7.12–7.15 (m, 1H, ArH), 7.23–7.34 (m, 5H, ArH), 7.50 (s, 1H, ArH); 13C NMR (75 MHz, CDCl3) δ = 29.8, 41.0, 52.1, 60.3, 111.8, 127.4, 127.8, 128.3, 128.6, 128.7, 128.9, 137.8, 144.6, 150.7, 171.5; mass (ES+) m/z = 302.1 (M++1); DART-HRMS calcd. for C16H16NO3S 302.0851; Found 302.0850.
:20, v/v); νmax (Neat) 1731 (CO2Me) cm−1; 1H NMR (200 MHz, CDCl3) δ = 2.75–2.88 (m, 1H, CH), 3.13 (dd, 1H, J1 = 18.8 Hz, J2 = 5.5 Hz, CH2), 3.45 (dd, 1H, J1 = 18.8 Hz, J2 = 12.9 Hz, CH2), 3.58 (s, 3H, OCH3), 5.24 (d, 1H, J = 6.8 Hz, CH), 6.45–6.48 (m, 1H, ArH), 6.93 (d, 1H, J = 3.4 Hz, ArH), 7.22–7.50 (m, 5H, ArH), 7.50 (d, 1H, J = 0.5 Hz, ArH); 13C NMR (75 MHz, CDCl3) δ = 25.6, 43.5, 52.3, 61.8, 111.3, 111.6, 127.5, 127.7, 127.9, 128.7, 141.8, 144.4, 149.0, 151.3, 172.7; mass (ES+) m/z = 302.1 (M++1); DART-HRMS calcd. for C16H16NO3S 302.0851; Found 302.0855.
:20, v/v); νmax (Neat) 1725 (CO2Me) cm−1; 1H NMR (200 MHz, CDCl3) δ = 2.33 (s, 3H, CH3), 3.02–3.07 (m, 1H, CH), 3.18–3.22 (m, 2H, CH2), 3.69 (s, 3H, CH3), 5.77 (d, 1H, J = 5.0 Hz, CH), 6.95 (s, 1H, ArH), 7.09 (brs, 4H, ArH), 7.17–7.22 (m, 2H, ArH), 7.30 (d, 2H, J = 2.0 Hz, ArH), 7.35 (brs, 6H, ArH); 13C NMR (50 MHz, CDCl3) δ = 21.5, 29.8, 41.2, 52.0, 60.8, 105.7, 125.6, 127.1, 127.9, 128.2, 128.5, 128.7, 129.0, 129.3, 138.1, 138.6, 140.0, 144.7, 151.4, 154.1, 171.9; mass (ES+) m/z = 468.2 (M++1); DART-HRMS calcd. for C28H26N3O2S 468.1746; Found 468.1750.
:20, v/v); νmax (Neat) 1725 (CO2Me) cm−1; 1H NMR (200 MHz, CDCl3) δ = 2.32 (s, 3H, CH3), 2.81–2.91 (m, 1H, CH), 3.14 (dd, 1H, J1 = 12.6 Hz, J2 = 3.6 Hz, CH2), 3.47 (dd, 1H, J1 = 18.8 Hz, J2 = 13.2 Hz, CH2), 3.59 (s, 3H, CH3), 5.27 (d, 1H, J = 7.0 Hz, CH), 6.96 (s, 1H, ArH), 7.08 (brs, 5H, ArH), 7.30 (brs, 2H, ArH), 7.34 (brs, 7H, ArH); 13C NMR (75 MHz, CDCl3) δ = 21.4, 25.7, 43.6, 52.3, 61.8, 105.8, 125.6, 127.2, 127.5, 127.6, 127.7, 127.9, 128.7, 129.0, 129.3, 138.6, 140.1, 142.0, 144.7, 173.0; mass (ES+) m/z = 468.2 (M++1); DART-HRMS calcd. for C28H26N3O2S 468.1746; Found 468.1747.
:20, v/v); νmax (Neat) 1727 (CO2Me) cm−1; 1H NMR (200 MHz, CDCl3) δ = 2.86–2.93 (m, 1H, CH), 3.20 (dd, 1H, J1 = 12.7 Hz, J2 = 3.6 Hz, CH2), 3.46–3.57 (m, 1H, CH2), 3.62 (s, 3H, OCH3), 5.34 (d, 1H, J = 6.5 Hz, CH), 7.21–7.23 (m, 1H, ArH), 7.25 (s, 1H, ArH), 7.30–7.42 (m, 6H, ArH), 7.47–7.52 (m, 1H, ArH), 7.72–7.77 (m, 1H, ArH); 13C NMR (50 MHz, CDCl3) δ = 25.4, 52.2, 52.5, 61.8, 104.6, 127.1, 127.2, 127.3, 127.7, 127.9, 128.4, 128.8, 130.5, 131.1, 131.2, 133.0, 137.3, 140.8, 161.5, 172.2; mass (ES+) m/z = 413.1 (M++1); DART-HRMS calcd. for C21H18ClN2O3S 413.0727; Found 413.0731.
:1).
69% as colorless oil (73 mg from 100 mg); Rf = 0.32 (hexanes:EtOAc, 80:20, v/v); νmax (Neat) 1735 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 1.30–1.36 (m, 3H, CH2), 1.46–1.58 (m, 4H, CH2), 1.62–1.68 (m, 3H, CH2), 1.79–2.06 (m, 10H,CH2 and CH), 2.27–2.38 (m, 2H, CH), 2.61–2.67 (m, 1H, CH2), 2.89–3.07 (m, 4H, CH and CH2), 3.32 (dd, 1H, J1 = 12.5 Hz, J2 = 8.8 Hz, CH2), 3.54 (s, 3H, CH3), 3.65 (s, 3H, CH3), 4.96 (d, 1H, J = 7.2 Hz, CH), 5.47 (d, 1H, J = 3.3 Hz, CH), 7.06–7.09 (m, 2H, ArH), 7.16–7.19 (m, 2H, ArH), 7.22–7.35 (m, 6H, ArH); 13C NMR (75 MHz, CDCl3) δ = 22.0, 22.8, 25.9, 26.2, 29.5, 29.8, 31.3, 31.4, 31.5, 31.6, 41.0, 43.4, 50.3, 50.6, 51.9, 52.1, 60.3, 61.3, 127.3, 127.5, 127.7, 128.0, 128.4, 128.6, 138.4, 142.0, 166.4, 167.0, 172.0, 173.1; mass (ES+) m/z = 318.1 (M++1); DART-HRMS calcd. for C18H24NO2S 318.1528; Found 318.1531.
:20, v/v); νmax (Neat) 1711 (CO2Me) cm−1; 1H NMR (200 MHz, CDCl3) δ = 2.34 (s, 3H, CH3), 3.87 (s, 3H, OCH3), 4.90 (d, 2H, J = 5.0 Hz, CH2), 7.10 (brs, 3H, ArH), 7.25 (s, 1H, ArH), 7.28–7.31 (m, 3H, ArH), 7.34–7.38 (m, 5H, ArH), 7.41–7.43 (m, 3H, ArH), 7.98 (s, 1H, CH), 9.04 (s, 1H, NH); 13C NMR (75 MHz, CDCl3) δ = 21.4, 42.8, 52.5, 110.6, 125.5, 126.4, 126.8, 128.1, 128.2, 128.6, 128.7, 128.8, 128.9, 129.1, 129.3, 129.7, 129.9, 134.1, 138.7, 139.7, 140.1, 144.8, 145.0, 151.1, 151.2, 168.0, 187.3; mass (ES+) m/z = 468.1 (M++1); DART-HRMS calcd. for C28H26N3O2S 468.1746; Found 468.1747.
:20, v/v); νmax (Neat) 1715 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 0.87–0.94 (m, 2H, CH2), 1.28–1.30 (m, 2H, CH2), 1.74–1.83 (m, 1H, CH), 3.88 (s, 3H, OCH3), 4.81 (d, 2H, J = 5.0 Hz, CH2), 7.30 (t, 1H, J = 3.9 Hz, ArH), 7.39–7.41 (m, 4H, ArH), 7.83 (brs, 1H, NH), 7.91 (s, 1H, CH); 13C NMR (50 MHz, CDCl3) δ = 12.1, 24.6, 43.5, 52.6, 126.2, 127.3, 128.1, 128.9, 129.8, 129.9, 133.9, 144.4, 168.4, 206.6; mass (ES+) m/z = 276.2 (M++1); DART-HRMS calcd. for C15H18NO2S 276.1058; Found 276.1060.
Acknowledgements
Two of the authors (SB and AM) gratefully acknowledge the financial support from Council of Scientific and Industrial Research, New Delhi in the form of fellowships. Authors gratefully acknowledge the SAIF Division of CDRI for recording all the spectroscopic and analytical data. This work was supported by a grant from Department of Science and Technology, New Delhi.References
- (a) W. Zhong, Y. Liu, G. Wang, L. Hong, Y. Chen, X. Chen, Y. Zheng, W. Zhang, W. Ma, Y. Shen and Y. Yao, Org. Prep. Proced. Int., 2011, 43, 1–66 CrossRef CAS; (b) D. Basavaiah, B. S. Reddy and S. S. Badsara, Chem. Rev., 2010, 110, 5447–5674 CrossRef CAS, and references cited therein (c) V. Declerck, J. Martinez and F. Lamaty, Chem. Rev., 2009, 109, 1–48 CrossRef CAS; (d) S. Gowrisankar, H. S. Lee, S. H. Kim, K. Y. Lee and J. N. Kim, Tetrahedron, 2009, 8769–8780 Search PubMed; (e) V. Singh and S. Batra, Tetrahedron, 2008, 64, 4511–4574 CrossRef CAS.
- (a) W. Zhong, W. Ma and Y. Liu, Tetrahedron, 2011, 67, 3509–3518 CrossRef CAS; (b) Z. Chen and D. L. J. Clive, J. Org. Chem., 2010, 75, 7014–7017 CrossRef CAS; (c) D. S. Yadav, R. Patel and V. P. Srivastava, Tetrahedron Lett., 2009, 50, 1335–1339 CrossRef; (d) M. M. Sa, L. Fernandes, M. Ferreira and A. J. Bortoluzzi, Tetrahedron Lett., 2008, 49, 1228–1232 CrossRef CAS.
- S. Bhowmik, A. Mishra and S. Batra, RSC Adv., 2011 10.1039/c1ra00399b.
- (a) N. Karodia In Comprehensive Heterocyclic Chemistry Vol. 4, 4.01 (ed. A. R. Katrizky, C. A. Ramsden, R. J. K. Taylor, E. F. V. Scriven) Pergamon: London, 2008, 568-604 Search PubMed; (b) A. Harrison-Marchand, D. Mauger, A. Guingant and J. P. Pradere, Tetrahedron, 2004, 60, 1827–1839 CrossRef CAS; (c) N. Leflemme, P. Dallemagne and S. Rault, Tetrahedron Lett., 2004, 45, 1503–1505 CrossRef CAS.
- T. Ozturk, E. Ertas and O. Mert, Chem. Rev., 2007, 107, 5210–5278 CrossRef CAS.
- From the mixture of products afforded from 6d, however it was possible to salvage (4R,5S)-4-(4-Fluorophenyl)-2-phenyl-5,6-dihydro-4H-1,3-thiazine-5-carbonitrile but in 3% yields only. The spectroscopic data for this compound is included in the supporting information†.
Footnotes |
| † Electronic Supplementary Information (ESI) available: Spectral data for remaining compounds and copies of 1H- and 13C-NMR spectra for all compounds, NOESY spectra for compounds syn-7d and anti-7d are included. See DOI: 10.1039/c1ra00362c/ |
| ‡ CDRI Communication no. 8107. |
| § Authors have equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2011 |