MgAl2O4 nanoparticles: A new low-density additive for accelerating thermal decomposition of ammonium perchlorate†
Xiangfeng
Guan
a,
Liping
Li
b,
Jing
Zheng
b and
Guangshe
Li
*a
aState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, 350002, P. R. China. E-mail: guangshe@ fjirsm.ac.cn; Fax: (+86) 591-83702122
bKey Laboratory of Optoelectronic Materials Chemistry and Physics, Chinese Academy of Sciences, Fuzhou, 350002, P. R. China
First published on 31st October 2011
Abstract
Developing low-density additives with high activities is significantly important for thermal decomposition of ammonium perchlorate and the relative technical applications. In this paper, nanoparticles of magnesium aluminate (MgAl2O4) with a low density of 3.02–3.17 g cm−3 were synthesized by a self-generated template path, in which citric acid was introduced to ensure homogeneous distribution of metal cations at atomic level and serve as the carbon source of the self-generated carbon template. Carbon template was generated by pyrolysis and carbonization of citrate after calcinations at 800 °C in N2, and then removed with annealing in air at high temperatures, resulting in porous MgAl2O4 with a specific surface area as high as 291 m2 g−1. Depending on the annealing temperatures, the primary sizes of MgAl2O4 nanoparticles that built up the porous structures were adjusted from 6.2 to 24.7 nm. The structural characteristics of MgAl2O4 nanoparticles were systematically studied by X-ray diffraction, transmission electron microscopy, Braun-Emmet-Teller analysis, and the catalytic role were evaluated by thermal decomposition of ammonium perchlorate. All samples were indicated to be X-ray-pure MgAl2O4, among which MgAl2O4 nanoparticles with a largest surface area of 291 m2 g−1 and pore volume of 0.24 cm3 g−1 was proved to have an optimum catalytic activity: exothermic peaks of AP thermal decomposition shifted towards the lower temperatures by 78.3 °C for high-temperature decomposition process. The relative kinetic process was also investigated.
1. Introduction
Catalyzed thermal decomposition of ammonium perchlorate (AP) plays a key role in the fields of aerospace, universal exploration, and satellite launching. This is because that AP is an important oxidizer in solid composite propellant for rockets, and its burning behavior greatly influences the ballistics of the rockets.1–3 The kernel of catalytic technologies for AP thermal decomposition is the additives, which can realize the highly efficient decomposition of AP at relatively low temperatures. Up to date, numerous additives (e.g.TiO2, CoO, Fe2O3, ZnO, CuO, and NiO)4–12 for AP thermal decomposition have been studied and some additives with high catalytic properties towards thermal decomposition of AP have been obtained. Nevertheless, these additives suffer from relatively high densities, which may bring the rocket extra load. Therefore, hunting for low-density additives with high catalytic activities is one of the future directions for AP thermal decomposition and the relevant research fields, and possibly to be a hot research topic in both materials and chemistry fields.By systematically analyzing the substantial literature data, we anticipate that MgAl2O4 has a high possibility to be a potential low-density additive with a high catalytic activity for AP thermal decomposition, which is based on the following considerations: (1) Metals Mg and Al are two typical elements with relatively low densities, the atomic numbers of which are 12 and 13, respectively. Additionally, the theoretical density of MgAl2O4 is as low as 3.58 g cm−3;13 (2) MgAl2O4 exhibits a very high thermal stability, and its melting point is as high as 2135 °C;14 (3) MgAl2O4 is a substance that has shown a catalytic role in many important reactions,15,16 like improving the dimerization reaction of cyclohexanone,15 or as SOx transfer catalyst for oxidation reaction of sulfur dioxide.16 However, up to date, there have not been enough evidences to prove that MgAl2O4 possesses a high catalytic role towards the thermal decomposition of AP.
Herein, we developed a novel self-generated template method to synthesize MgAl2O4 nanoparticles. The specific surface area, porous structure, and primary size of these MgAl2O4 nanoparticles were tailored by removing the self-generated carbon template and subsequent high-temperature annealing. All MgAl2O4 samples were demonstrated to have an apparent catalytic role towards the thermal decomposition of AP.
2. Experimental details
2.1. Sample preparation
We proposed a self-generated template method to prepare MgAl2O4 samples, which can be designed in term of three steps. As illustrated in Fig. 1, the first step involves the formation of a dried gel via a solution chemistry, in which citric acid plays dual roles: Ensuring the homogeneous distribution of metal cations at atomic level, and serving as the carbon source of the self-generated template. The second step is the in situ formation of carbon template, which was generated by pyrolysis and carbonization of citrate after calcinations at 800 °C in N2. The third step involves the formation of MgAl2O4 nanoparticles with porous structure, which was achieved by removing the carbon template in air at high temperatures.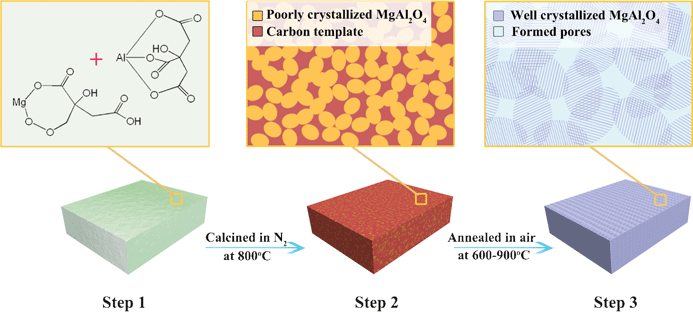 | ||
| Fig. 1 Scheme that illustrates the self-generated template approach to porous MgAl2O4. Step 1: formation of dry gel that contains the homogenous mixed metal citrate precursor after solvent evaporation; step 2: formation of amorphous carbons template around the poorly crystallized MgAl2O4 by pyrolysis of citrate; and step 3: formation of well crystallized MgAl2O4 with porous structure by removing the template. | ||
The typical synthesis procedure to the samples can be described as follows: Initially, analytical grade chemicals of Mg(NO3)2·6H2O (≥ 99.0%), Al(NO3)3·9H2O (≥ 99.0%), and citric acid (C6H8O7·H2O) (≥ 99.5%) were used as the starting materials without further purification. Secondly, 0.01 molar Mg(NO3)2·6H2O and 0.02 molar Al(NO3)3·9H2O were dissolved in 100 ml deionized water to form a transparent solution. Then, citric acid was added into the above solution with a molar ratio of citric acid to metal ions at 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. A lightly yellow transparent solution was formed after heating in 70 °C water-bath for several hours with an increased viscosity, resulting in the formation of yellow gels. The resultant gels were dried at 150 °C for 2 h to obtain a fluffy dry gel, the citrate precursor of MgAl2O4. After grinding, the precursor was heated in N2 at 800 °C for 1 h, which gives rise to a black amorphous mixture. Ultimately, the resulting mixture was annealed in air at 600–900 °C for 1 h to obtain the final samples MgAl2O4. The sample before annealing was named as S-P, while that after annealing at 600, 700, 800, and 900 °C were named as S-600, S-700, S-800, and S-900, respectively.
:1. A lightly yellow transparent solution was formed after heating in 70 °C water-bath for several hours with an increased viscosity, resulting in the formation of yellow gels. The resultant gels were dried at 150 °C for 2 h to obtain a fluffy dry gel, the citrate precursor of MgAl2O4. After grinding, the precursor was heated in N2 at 800 °C for 1 h, which gives rise to a black amorphous mixture. Ultimately, the resulting mixture was annealed in air at 600–900 °C for 1 h to obtain the final samples MgAl2O4. The sample before annealing was named as S-P, while that after annealing at 600, 700, 800, and 900 °C were named as S-600, S-700, S-800, and S-900, respectively.
It should be mentioned that the present self-generated template method is superior to the well-established Pechini methods in preparing porous structure, since when using the latter methods,17–19 the precursor gel were always directly calcined in air, which does not facilitate the formation of carbon template necessary for porous structure.
2.2. Sample characterization
Phase structures of the samples were characterized by X-ray diffraction (XRD) on a Rigaku MiniFlex II X-ray diffractometer using a copper target. Crystallite sizes were calculated using the Scherrer formula as follows:| D = 0.9λ/(β cosθ) | (1) |
Specific surface areas of the samples were measured by N2 adsorption isotherms at 77 K on a Micromeritics ASAP 2000 surface area and porosity analyzer after degassing the samples in a flowing nitrogen atmosphere at 120 °C for 12 h in a separate degassing unit attached to the instrument. The actual densities of the samples were measured by a pycnometer method20 using pure water as the immersion liquid.
Catalytic roles of the samples towards thermal decomposition of AP were studied by differential scanning calorimeter (DSC) using DTA404PC thermal analyzer at a heating rate of 15 °C min−1 in N2 over a temperature range of 30–600 °C. DSC experiments were performed using an open alumina crucible. AP and the samples were mixed at a mass ratio of 98:2 for thermal decomposition analyses. A mixture with a total mass of 5 mg was used in all runs.
3. Results and discussion
3.1 Density comparison of the additives for AP thermal decomposition
Table 1 lists the density data of the additives reported in literature for AP thermal decomposition, along with other properties including melting point and catalytic roles. To emphasize the importance and possibility of MgAl2O4 as a new additive with low-density and high thermal stability, the relevant physical properties for MgAl2O4 spinel is also shown. From Table 1, it can be seen that the additives reported for AP thermal decomposition can be classified as three types: metals, metal oxides, and metal hydrides. For the metallic additives, both Ni and Co exhibit the relatively high thermal stability because of the high melting points (>1400 °C), while their densities are also relatively high, >8.9 g cm−3. Although the density of Al is lower than those of all metal oxides, the melting point of Al is only 660 °C. Metal hydride MgH2 has the lowest density among all listed materials, while its application is limited by its poor thermal stability due to its low melting points of 327 °C. Comparatively, metal oxides can be the promising additives. The relevant additives reported to have a melting point higher than 2000 °C include CeO2, NiO, and MgO, in which the densities of the former two are higher than 7 g cm−3, while that for the latter one is only 3.65 g cm−3. From Table 1, it is clear that among all metal oxides materials, MgAl2O4 possesses a very low density of 3.58 g cm−3 and a high melting point of 2135 °C. Therefore, MgAl2O4 is of the greatest possibility to be a new low-density additive for accelerating the thermal decomposition of AP.| Materials | Densitya (g cm−3) | Melting pointa (°C) | Catalytic role | Materials | Density (g cm−3) | Melting point (°C) | Catalytic role |
|---|---|---|---|---|---|---|---|
| a The data of density and melting point without specific citation are referred to ref. 42. | |||||||
| Ni | 8.91 | 1453 | √ (ref. 21) | Cu2O | 6.04 | 1235 | √ (ref. 32) |
| Co | 8.90 | 1494 | √ (ref. 22) | ZnO | 5.60 | 1975 | √ (ref. 8,9) |
| CdO | 8.15 | 1540 | √ (ref. 23) | α-Fe2O3 | 5.25 | 1565 | √ (ref. 6,7) |
| CeO2 | 7.65 | 2400 | √ (ref. 24) | CoFe2O4 | 5.20 33 | 1567 34 | √ (ref. 35) |
| NiO | 7.45 | 2000 | √ (ref. 25) | MnO2 | 5.08 | 530 | √ (ref. 36) |
| LaCoO3 | 7.30 26 | 1740 27 | √ (ref. 28) | MoO3 | 4.69 | 801 | √ (ref. 37) |
| Nd2O3 | 7.28 | 1900 | √ (ref. 29) | MgO | 3.65 | 2800 | √ (ref. 38) |
| CuO | 6.32 | 1450 | √ (ref. 30) | MgAl2O4 | 3.58 13 | 2135 14 | ? |
| CoO | 6.44 | 1935 | √ (ref. 5) | Al | 2.70 | 660 | √ (ref. 39) |
| Co3O4 | 6.07 | 900 | √ (ref. 31) | MgH2 | 1.45 | 327 40 | √ (ref. 41) |
In the following, we used a self-generated template strategy to prepare porous MgAl2O4 nanoparticles and further examined their catalytic role in thermal decomposition of AP.
3.2 Optimized conditions to the formation of MgAl2O4
The formation conditions to MgAl2O4 were optimized by tuning the annealing temperatures. Fig. 2 shows XRD patterns of the samples before and after annealing in air. As indicated in Fig. 2, the sample S-P before annealing in air showed several broad and dispersed diffraction peaks, which indicates a poor crystalline character. After annealing at 600 °C in air, the diffraction peaks for S-600 became stronger in intensity. The mean crystallite size of S-600 was calculated to be about 6.2 nm using Scherrer formula for the peak (311). When the annealing temperature increased to 700 °C, the diffraction intensity was further increased, and the peak widths became narrowed. The mean crystalline size of S-700 was calculated to be 9.4 nm. All diffraction peaks for S-700 matched well the standard data for MgAl2O4 (JCPDS, No. 73–1959), while no component phases like MgO and Al2O3 are detected, which indicates the formation of single-phase spinel MgAl2O4. These spinel MgAl2O4 nanoparticles were well crystallized, as supported by the following TEM observations. This spinel structure was retained with increasing the annealing temperatures to 900 °C, while the crystalline size increased to 11.5 nm for S-800 and 24.7 nm for S-900.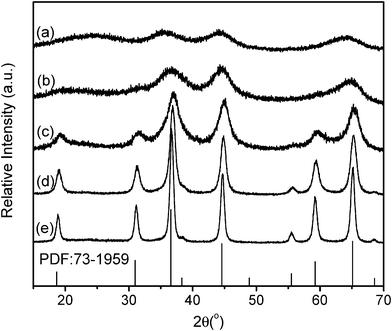 | ||
| Fig. 2 X-ray diffraction patterns of the samples: (a) S-P, (b) S-600, (c) S-700, (d) S-800, and (e) S-900. | ||
Since Mg and Al may form spinel structure within a certain compositional range, we carried out a detailed study on the chemical compositions of the samples by elemental mapping and quantitative analysis using EDS. As indicated by the elemental mapping results in Fig. 3 and S1†, both elements Mg and Al are homogeneously distributed in all samples, which further confirms the single-phase character of the samples. The results of quantitative elemental analysis are summarized in Table 2. It can be seen that the atomic ratio of Mg to Al is approximately 1:2, closer to the stoichiometry of MgAl2O4. Therefore, using the present preparation method, MgAl2O4 spinel was formed after annealing at 600 °C, and higher temperature annealing did not significantly affect the compositions of MgAl2O4.
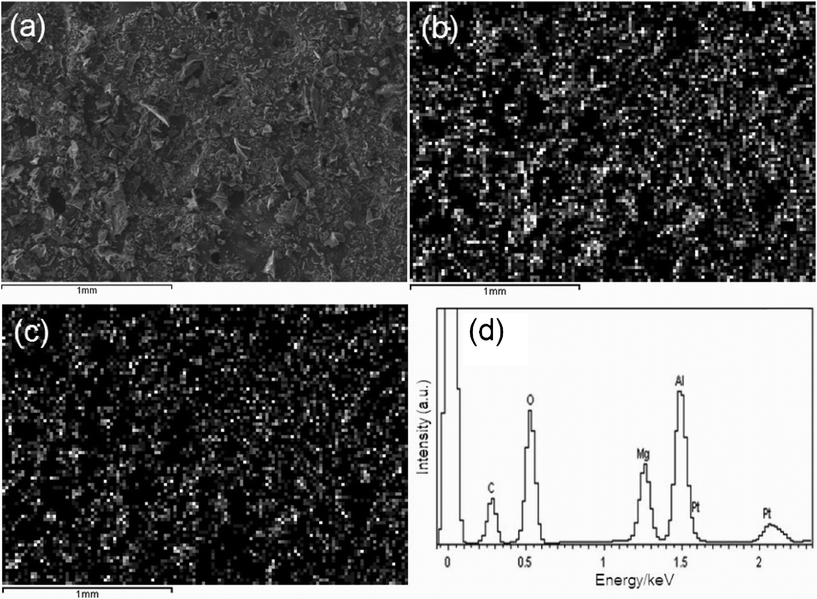 | ||
| Fig. 3 SEM image (a) and X-ray mapping of Mg (b) and Al (c), and EDS spectrum (d) of S-700. | ||
| Sample | Chemical compositionsa | Specific surface area (m2 g−1) | Pore volume (cm3 g−1) | Actual density (g cm−3) | ||
|---|---|---|---|---|---|---|
| Mg | Al | Mg/Al ratio | ||||
| a Chemical compositions were obtained by EDS measurements. | ||||||
| S-600 | 0.142 | 0.287 | 1:2 |
150 | 0.16 | 3.04 |
| S-700 | 0.144 | 0.285 | 1:2 |
291 | 0.24 | 3.02 |
| S-800 | 0.144 | 0.285 | 1:2 |
91 | 0.15 | 3.17 |
| S-900 | 0.143 | 0.286 | 1:2 |
40 | 0.13 | 3.15 |
3.3 Construction of porous MgAl2O4
Fig. 4a shows the adsorption–desorption isothermals of the samples. It can be seen that all single-phase MgAl2O4 exhibited a porous structure. For S-600, according to the IUPAC classification,43 the isotherms are qualitatively of a type IV shape, which is characteristic of porous structures.44,45 The hysteresis is of type H2, typical for capillary condensation inside the ink-bottle shaped mesopores. With increasing the annealing temperatures, the isotherms remain a type IV shape, while the hysteresis tends towards a H1 type, which indicates a greater regularity of the cross section along the longitudinal direction of the pores.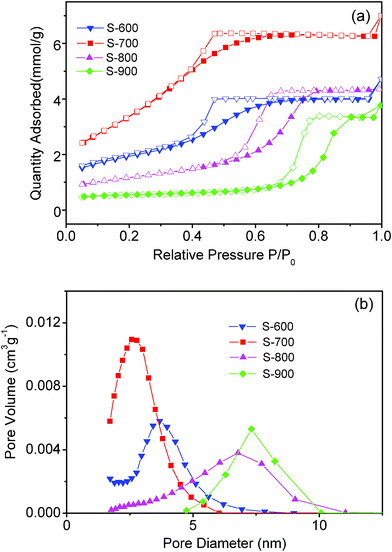 | ||
| Fig. 4 (a) Adsorption–desorption isothermals and (b) BJH adsorption size distribution of the samples. | ||
Generally, pore size distribution can be obtained from both adsorption and desorption branches of the isotherm data. It is well established46 that in the case of the isotherm of type IV accompanied by a type H2 hysteresis loop, the pore size distribution derived from the desorption branch is often greatly affected by a tensile strength effect (TSE), which may lead to a narrow pore size distribution, not reflecting the real porous structures. To get insight into the impact of TSE on the pore size distribution, Barrett–Joyner–Halenda (BJH) adsorption and desorption size distribution of all samples were measured, as shown in Fig. 4b and S2†. BJH adsorption pore size distribution (Fig. 4b) indicates that all samples were mesoporous. The pore size distribution became broader as the annealing temperatures increased from 600 to 900 °C, while the maximum pore diameter shifted from 3.6 to 7.3 nm.
Such cases were apparently altered when the pore size distribution is derived from the desorption branch of the isotherm data. As shown in S2†, the pore size distribution for all samples became narrower and the maximum pore diameters were apparently changed: The maximum pore diameter for S-700 increased from 2.6 to 3.2 nm, while that for S-800 decreased from 6.7 to 4.7 nm, which are in accordance with the previous literature data where a narrow distribution of pores centered at 4 nm occurs due to TSE phenomenon.46 Therefore, to avoid the impact of TSE, pore size distribution of the samples was derived from the adsorption branch of the isotherm. The total pore volume and specific surface areas for these porous samples are summarized in Table 2. It can be seen that S-700 has a largest total pore volume of 0.24 cm3 g−1 and a highest specific surface areas of 291 m2 g−1. All these porous MgAl2O4 nanoparticles also showed low densities experimentally measured (Table 2).
Porous structures of all MgAl2O4 samples were confirmed by TEM observations. As shown in S3†, all MgAl2O4 samples were composed of many small nanoparticles, which are cross-linked with each other to form a porous structure. For S-600, the pores having a diameter of about 4 nm were randomly arranged but distributed homogeneously, as indicated by the light regions in Fig. 5a. The pore walls were constructed by many crystallized MgAl2O4 nanoparticles that are randomly oriented and overlapped from each other. These nanoparticles had an average size around 6 nm in good agreement with the mean crystal size of 6.2 nm calculated from XRD peak broadening. It is also noted that there are some amorphous portions without long-range ordering for S-600, which became less with the increase of the annealing temperature.
 | ||
| Fig. 5 HRTEM images of the samples: (a) S-600, (b) S-700, (c) S-800, (d) S-900. Typical pores are sketched in red. | ||
With the annealing temperature increased to 700 °C, as shown in Fig. 5b, the pore structure of S-700 is similar to that of S-600 except that the pore size decreased to about 3 nm and the average size of the nanoparticles on the pore walls increased to about 9 nm. Such a pore structure retained up to an annealing temperature of 900 °C (Fig. 5c, d), while the pore size and crystalline size of nanoparticles were increased. It is consistent with the trend obtained from BJH adsorption size distribution and XRD measurements.
Surface chemistry of all samples was studied by FTIR. As shown in S4†, all MgAl2O4 samples showed similar infrared spectra, in which OH− bonding characteristic of surface hydration layer was apparent. Therefore, all MgAl2O4 samples were terminated by surface hydration layers, closely related to the porous nature.
3.4 Thermal decomposition of AP in the presence of porous MgAl2O4
The heat released during AP decomposition was kinetically measured by DSC in a flowing nitrogen gas. As indicated in Fig. 6, all porous MgAl2O4 nanoparticles exhibited an apparent catalytic role towards the thermal decomposition of AP. With no addition of the porous MgAl2O4 nanoparticles, two endothermic peaks and one exothermic peak were observed: The first endothermic peak maximized at about 247.7 °C corresponds to a phase transition of AP from the orthorhombic to cubic,47 in which no mass changes appeared in the corresponding temperature range. The second exothermic peak located at about 331.9 °C (Ta as marked in Fig. 6), corresponds to the low-temperature decomposition (LTD) process. The third endothermic peak maximized at 437.8 °C (Tb as shown in Fig. 6) is referred to the high-temperature decomposition (HTD) process.48 Such cases were apparently altered when the porous MgAl2O4 nanoparticles were added to AP. As shown in Fig. 6, with the addition of MgAl2O4 nanoparticles, the decomposition temperatures of HTD process were all shifted: For S-600, the peak of HTD stage was exothermic, and the peak temperature was at 387.1 °C. For S-700, the peak temperature of HTD stage moved downwards to 359.5 °C, while the peak became stronger in intensity and narrower in width. For S-800, the peak temperature of HTD stage moved to 401.6 °C and became weaker in intensity. Further increasing the annealing temperature up to 900 °C, the peak temperature of HTD stage of AP was at 431.2 °C, while the HTD peak was changed to an endothermic process. Therefore, S-700 is proved to possess an optimum catalytic role in thermal decomposition of AP.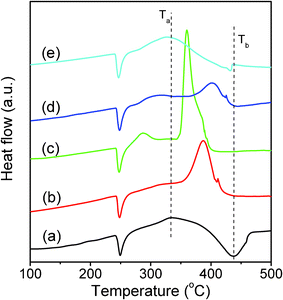 | ||
| Fig. 6 DSC curves for AP (a) and that mixed with additives of S-600 (b), S-700 (c), S-800 (d), and S-900 (e). | ||
Having these experimental results in mind, one question appeared: What is the primary reason for the optimum catalytic role of S-700? To answer this question, one has to take into account several factors that may contribute to the thermal decomposition of AP. For instance, particle size effect is popularly considered to be crucial for the catalytic decomposition of AP. For the present work, particle size effect can be first dismissed, since it cannot rationalize why S-700 with a particle size of 9.4 nm offers a higher performance than S-600 (with a smaller particle size of 6.2 nm) or S-800 and S-900 (with larger particle sizes of 11.5 nm and 24.7 nm, respectively). Alternatively, specific surface area and pore volume can be the primary reasons for the catalytic role, since more reactive sites can be generated in porous MgAl2O4 nanoparticles necessary for accelerating AP decomposition. Although the interactions between the porous MgAl2O4 nanoparticles and AP are still not fully understood due to the complexity of AP decomposition process, we believe that porous MgAl2O4 nanoparticles may accelerate ammonia oxidation and ClO4− species dissociation, the intermediates during the AP decomposition. Among all porous MgAl2O4 nanoparticles, S-700 showed a highest surface area and largest pore volume (Table 2), which explains its best performance in accelerating AP decomposition.
In addition to these factors, amorphous portion of the samples may contribute to the catalytic role, since in most cases, amorphous materials can be reactive.49–51 For the present work, XRD and TEM measurements have demonstrated that the crystallinity of porous nanoparticles increased with the annealing temperature. Namely, the amorphous portion decreased with increasing the annealing temperature from 600 to 900 °C, which does not follow the sequence of the catalytic roles of the corresponding samples. For example, S-700 offers a highest catalytic activity among all samples, in which amorphous material is apparently less than that for S-600. Therefore, it is reasonable that crystalline wall of the porous MgAl2O4 nanoparticles is likely the primary part of the catalytic role.
S-700 was chosen as the additive to investigate the kinetic process of the thermal decomposition of AP, focusing on the activation energy of HTD stage. It is well established52 that the activation energy for the thermal decomposition of AP can be obtained by the following relationship between decomposition temperature and heating rate:
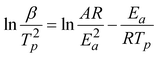 | (2) |
Fig. 7a shows the DSC curves of the mixtures of AP with additives of S-700 at given heating rates. When the heating rate is 5 K min−1, there is a narrow exothermic peak in HTD stage. With an increase in heating rate, the HTD peak was widened and the peak temperature shifted from 354.0 °C at 5 K min−1 to 365.1 °C at 30 K min−1. Fig. 7b shows the experimentally measured ln(β/Tp2) versus 1/Tp with and without additives of S-700. As can be seen, for pure AP, the activation energy of HTD was calculated to be 108 kJ mol−1, which is close to the value previously reported in literature.53 In the presence of S-700, the activation energy of AP decomposition became as large as 483 kJ mol−1. It appears that adding S-700 increases, rather than decreases the activation energy (energy barrier) of the AP decomposition reaction, we may conclude that S-700 is an effective additive, though it may also catalyze the high-temperature decomposition of ammonium perchlorate as anatase does.4
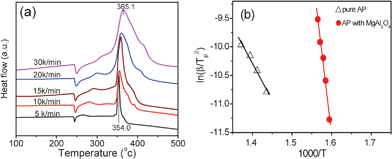 | ||
| Fig. 7 (a) DSC curves of AP mixed with S-700 at given heating rates, and (b) dependence of ln(β/Tp2) on 1/Tp for AP decomposition with and without additive S-700. Scattered points are corresponding to the experimental data, while lines denote the data fit results. | ||
4. Conclusions
This paper reports our findings on a new low-density additive, porous MgAl2O4 nanoparticles, towards thermal decomposition of ammonium perchlorate. The main results can be summarized as follows:(1) MgAl2O4 nanoparticles were synthesized by developing a self-generated template method. Using this method, MgAl2O4 nanoparticles were achieved to be porous with pore walls being constructed by randomly oriented spinel nanoparticles.
(2) The pore volume of the porous MgAl2O4 nanoparticles was tailored from 0.13 to 0.24 cm3 g−1 by removing the self-generated carbon template after high-temperature annealing in air.
(3) When acting as the additives, all porous MgAl2O4 nanoparticles catalyzed the high-temperature decomposition of ammonium perchlorate, a key component of solid propellant. An optimum catalyzed AP decomposition was obtained when porous MgAl2O4 nanoparticles were controlled to have a largest surface area of 291 m2 g−1 and pore volume of 0.24 cm3 g−1.
The findings reported in this work are significantly important, which may help to explore more new additives towards thermal decomposition of ammonium perchlorate.
Acknowledgements
This work was financially supported by NSFC (No. 21025104, 91022018, 50972143), National Basic Research Program of China (2011CBA00501), Directional program CAS (KJCXZ-YW-M10), and FJIRSM fund (No. 2010KL002).References
- S. Vyazovkin and C. A. Wight, Chem. Mater., 1999, 11, 3386 CrossRef CAS
.
- L. Bereczki, K. Marthi, P. Huszthy and G. Pokol, J. Therm. Anal. Calorim., 2004, 78, 449 CrossRef CAS
- H. Xu, X. B. Wang and L. Z. Zhang, Powder Technol., 2008, 185, 176 CrossRef CAS
- D. L. Reid, A. E. Russo, R. V. Carro, M. A. Stephens, A. R. LePage, T. C. Spalding, E. L. Petersen and S. Seal, Nano Lett., 2007, 7, 2157 CrossRef CAS
- L. P. Li, X. F. Sun, X. Q. Qiu, J. X. Xu and G. S. Li, Inorg. Chem., 2008, 47, 8839 CrossRef CAS
- Y. F. Zhang, X. H. Liu, J. R. Nie, L. Yu, Y. L. Zhong and C. Huang, J. Solid State Chem., 2011, 184, 387 CrossRef CAS
- Z. Y. Ma, F. S. Li and H. P. Bai, Propellants, Explos., Pyrotech., 2006, 31, 447 CrossRef CAS
- X. F. Sun, X. Q. Qiu, L. P. Li and G. S. Li, Inorg. Chem., 2008, 47, 4146 CrossRef CAS
- J. Z. Yin, Q. Y. Lu, Z. N. Yu, J. J. Wang, H. Pang and F. Gao, Cryst. Growth Des., 2010, 10, 40 CAS
- J. W. Zhu, G. Y. Zeng, F. D. Nie, X. M. Xu, S. Chen, Q. F. Han and X. Wang, Nanoscale, 2010, 2, 988 RSC
- Y. P. Wang, J. W. Zhu, X. J. Yang, L. D. Lu and X. Wang, Thermochim. Acta, 2005, 437, 106 CrossRef CAS
- L. J. Chen, G. S. Li, P. Qi and L. P. Li, J. Therm. Anal. Calorim., 2008, 92, 765 CrossRef CAS
- L. R. Ping, A. M. Azad and T. W. Dung, Mater. Res. Bull., 2001, 36, 1417 CrossRef
- R. Lanos and R. Lazau, Mater. Chem. Phys., 2009, 115, 645 CrossRef
- R. S. Li, X. K. Wang, L. M. Qian and X. F. Qin, J. Fuel Chem. Tech., 1995, 23, 144 CAS
- M. Waqif, O. Saur, J. C. Lavalley, Y. Wang and B. A. Morrow, Appl. Catal., 1991, 71, 319 CrossRef CAS
- L. Macalik, P. E. Tomaszewski, R. Lisiecki and J. Hanuza, J. Solid State Chem., 2008, 181, 2591 CrossRef CAS
- Y. S. Chang, F. M. Huang, Y. Y. Tsai and L. G. Teoh, J. Lumin., 2009, 129, 1181 CrossRef CAS
- M. Naderi, A. Shamirian and M. Edrisi, J. Sol-Gel Sci. Technol., 2011, 58, 557 CrossRef CAS
- C. R. Jensen, R. J. Luxmoore, S. D. Vangundy and L. H. Stolzy, Agron. J., 1969, 61, 474 CrossRef
- X. J. Shen, J. P. Yang, Y. Liu, Y. S. Luo and S. Y. Fu, New J. Chem., 2011, 35, 1403 RSC
- Z. T. Liu, X. Li, Z. W. Liu and J. Lu, Powder Technol., 2009, 189, 514 CrossRef CAS
- G. Singh, I. P. S. Kapoor, R. Dubey and P. Srivastava, Mater. Sci. Eng. B-Adv. Funct. Solid-State Mater., 2011, 176, 121 CAS
- D. E. Zhang, A. M. Chen, M. Y. Wang, J. Y. Gong, X. B. Zhang, S. Z. Li, G. Q. Han, A. L. Ying and Z. W. Tong, Funct. Mater. Lett., 2011, 4, 97 CrossRef CAS
- J. J. Wang, C. Z. Wei, H. Pang, F. Gao, J. Z. Yin, L. N. Guan and Q. Y. Lu, Catal. Commun., 2011, 12, 1031 CrossRef CAS
- Z. Y. Zhang, J. Koppensteiner, W. Schranz, D. Prabhakaran and M. A. Carpenter, J. Phys.: Condens. Matter, 2011, 23, 145401 CrossRef
- N. Orlovskaya, Y. Gogotsi, M. Reece, B. Cheng and I. Gibson, Acta Mater., 2002, 50, 715 CrossRef CAS
- Y. P. Wang, X. J. Yang, L. D. Lu and X. Wang, Thermochim. Acta, 2006, 443, 225 CrossRef CAS
- D. V. Survase, M. Gupta and S. N. Asthana, Prog. Cryst. Growth Charact. Mater., 2002, 45, 161 CrossRef CAS
- L. J. Chen, L. P. Li and G. S. Li, J. Alloys Compd., 2008, 464, 532 CrossRef CAS
- X. F. Guan, G. S. Li, L. H. Zhou, L. P. Li and X. Q. Qiu, Chem. Lett., 2009, 38, 280 CrossRef CAS
- J. W. Zhu, H. Q. Chen, B. Xie, X. J. Yang, L. Lu and X. Wang, Chin. J. Catal., 2004, 25, 637 CAS
- C. Cannas, A. Falqui, A. Musinu, D. Peddis and G. Piccaluga, J. Nanopart. Res., 2006, 8, 255 CrossRef CAS
- G. Srinivasan, E. T. Rasmussen, J. Gallegos and R. Srinivasan, Phys. Rev. B: Condens. Matter, 2001, 64, 214408 CrossRef
- S. S. Zhao and D. X. Ma, J. Nanomater., 2010, 842816 Search PubMed
- K. Kishore, V. R. P. Verneker and M. R. Sunitha, J. Appl. Chem. Biotechnol., 1977, 27, 415 CAS
- S. A. Halawy and M. A. Mohamed, Collect. Czech. Chem. Commun., 1994, 59, 2253 CrossRef CAS
- G. R. Duan, X. J. Yang, J. Chen, G. H. Huang, L. D. Lu and X. Wang, Powder Technol., 2007, 172, 27 CrossRef CAS
- K. Jayaraman, S. R. Chakravathy and R. Sarathi, Combust., Explos. Shock Waves, 2010, 46, 21 CrossRef
- Y. Yoo, M. Tuck, R. Kondakindi, C. Y. Seo, Z. Dehouche and K. Belkacemi, J. Alloys Compd., 2007, 446, 84 CrossRef
- L. L. Liu, F. S. Li, C. L. Zhi, H. C. Song, Y. Yang and Q. S. Zhang, Rare Metal Mat. Eng., 2010, 39, 1289 CAS
-
J. A. Dean, Lange's Handbook of Chemistry, McGraw-Hill Book Company, New York, 15th edition 1999section 3.13 Search PubMed
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603 CrossRef CAS
- S. B. Wang and G. Q. Lu, Ind. Eng. Chem. Res., 1999, 38, 2615 CrossRef CAS
- J. J. Guo, H. Lou, H. Zhao, X. G. Wang and X. M. Zheng, Mater. Lett., 2004, 58, 1920 CrossRef CAS
- J. C. Groen, L. A. A. Peffer and J. Perez-Ramirez, Microporous Mesoporous Mater., 2003, 60, 1 CrossRef CAS
- V. V. Boldyrev, Thermochim. Acta, 2006, 443, 1 CrossRef CAS
- P. M. W. Jacobs and G. S. Persone, Combust. Flame, 1969, 13, 419 CrossRef CAS
- F. Adam and A. Lqbal, Microporous Mesoporous Mater., 2011, 141, 119 CrossRef CAS
- A. Molnar, Appl. Surf. Sci., 2011, 257, 8151 CrossRef CAS
- W. Y. Wang, Y. Q. Yang, H. A. Luo, H. Z. Peng, B. He and W. Y. Liu, Catal. Commun., 2011, 12, 1275 CrossRef CAS
- S. Morisaki and K. Komamiya, Thermochim. Acta, 1975, 12, 239 CrossRef CAS
- J. C. Oxley, J. L. Smith and B. R. Valenzuela, J. Energ. Mater., 1995, 13, 57 CrossRef CAS
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c1ra00489a/ |
| This journal is © The Royal Society of Chemistry 2011 |