Mechanism of the gold-catalyzed cyclopropanation of alkenes with 1,6-enynes†
Patricia
Pérez-Galán
,
Elena
Herrero-Gómez
,
Daniel T.
Hog
,
Nolwenn J. A.
Martin
,
Feliu
Maseras‡
and
Antonio M.
Echavarren§
*
Institute of Chemical Research of Catalonia (ICIQ), Av. Països Catalans 16, 43007, Tarragona, Spain. E-mail: aechavarren@iciq.es
First published on 21st September 2010
Abstract
The gold(I)-catalyzed intermolecular cyclopropanation of alkenes with 1,6-enynes is an electrophilic process, which is mechanistically related to the well-known Simmons–Smith reaction proceeding through zinc carbenoids. This cyclopropanation is stereospecific, even in cases where the reaction was found to proceed stepwise.
Introduction
Reactions of 1,6-enynes 1 with electrophilic metal complexes as catalysts have been extensively studied in the last few years, which has allowed for determination of the main pathways that follow coordination of the metal to the triple bond of these substrates.1,2 For gold(I)-catalyzed reactions, based on DFT calculations we have proposed that the initial η2-complexes 2 form anti-cyclopropyl gold(I) carbenes3 in a 5-exo-dig process that competes with the relatively less common 6-endo-digcyclizationvia metal carbenes4 (Scheme 1).3,4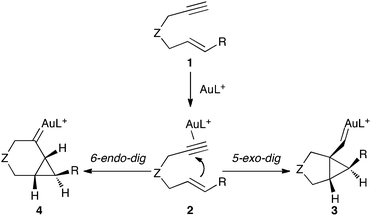 | ||
| Scheme 1 Major pathways in the gold(I)-catalyzed cyclization of 1,6-enynes. | ||
Intermediates 2 give rise to products of skeletal rearrangement,5,6 whereas in the presence of hetero- or carbonucleophiles, products of anti-addition are formed in a stereospecific process.3,7,8,9,10,11,12
Cyclopropyl metal carbenes were first proposed as intermediates in palladium(II)-catalyzed cyclizations of 1,6-enynes in the presence of 1,3-dienes.13 Experimental evidence for the formation of the corresponding gold(I) intermediates 3 was obtained in their intramolecular trapping with alkenes to give the corresponding cyclopropanes.3a,14,15 Similar results were previously reported for ruthenium(II)16 and platinum(II)-catalyzed reactions.17,18Gold(I) intermediates 3 have also been trapped with Ph2SO to form aldehydes.8f
Reaction of enynes1 with norbornene led to biscyclopropanes 5 by the intermolecular trapping of 3 with the alkene (Scheme 2).19Enynes with terminally unsubstituted alkenes (1, R = H) react via rearranged intermediate 6 to give cyclopropane7.
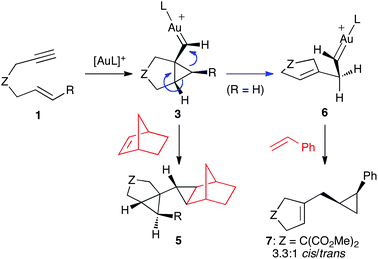 | ||
| Scheme 2 Trapping of intermediates 3 and 6 with alkenes. | ||
Propargyl carboxylates such as 8a–b also react stereoselectively with alkenes in the presence of gold(I) catalysts to give cyclopropanes9a–b with moderate to excellent cis-stereoselectivity (Scheme 3).20,21,22 This reaction, which is stereospecific with respect to the alkene,20,23,24 proceeds by a 1,2-carboxylate shift to form 10via transition state TS10–910–9 in which the interaction between the alkene substituent and the AuL fragment is minimized.20
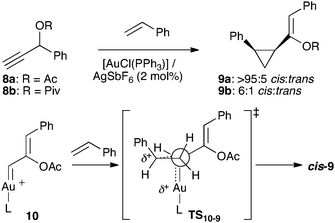 | ||
| Scheme 3 Gold(I)-catalyzed intermolecular cyclopropanation with propargyl carboxylates. | ||
The electrophilicity of the gold(I) complexes increase as the donor ability of the ligands decrease. Therefore, cationic gold(I) complexes show increased electrophilicity in the following order: N-heterocyclic carbene (A, A′, and B)11b < phosphine (C and D)10a,25 < phosphite (E and E′)11b (Fig. 1). Thus, for example, the addition of 1,3-diphenylpropane-1,3-dione to enyne1c leads to adducts 11 and 12 (Scheme 4) depending on the nature of the ligand on gold, reflecting the dual character, carbene-like (3) or carbocationic (3′), of the intermediates.11b,26 Similar arguments have been proposed in reactions of gold carbenes generated by the 1,2-acyl migration from propargyl carboxylates.23
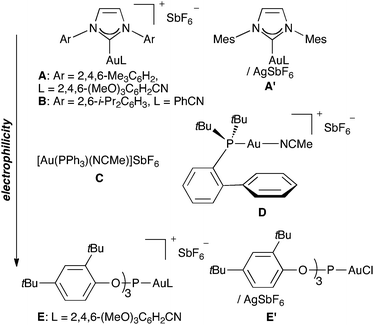 | ||
| Fig. 1 Gold(I)-complexes arranged in order of increasing electrophilicity (top to bottom). | ||
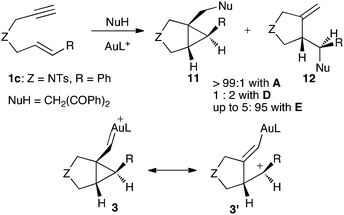 | ||
| Scheme 4 Gold(I)-catalyzed site-selective nucleophilic attack of a carbon nucleophile to enyne1c. | ||
The existence of 3 as discrete reactive intermediates in cyclizations of enynes has been questioned.9,27,28 Thus, based on similarities between gold(I)-catalyzed enyne cycloisomerizations and cationic polyene cyclizations, it was argued that the involved intermediates should be more appropriately described as gold-stabilized carbocations rather than gold carbenes.9 Furthermore, formation of cyclopropanes in these reactions was considered mechanistically ambiguous27 since cyclopropane formation can also occur in a stepwise, non-concerted process.29 Indeed, recent theoretical calculations suggest that cyclopropanation of electron-rich alkenes by gold carbenes is a stepwise process that occurs via cationic intermediates.30
We have shown that in extreme cases when the terminal substituent, R, at the alkene is strongly electron-releasing, reactions such as skeletal rearrangements and nucleophilic additions are no longer stereospecific and the reactive intermediates can be more consistently described as open carbocations such as 3′.31
Whereas cis-cyclopropanes were preferentially obtained in the reaction of gold(I)-catalyzed reaction of propargyl carboxylates with alkenes (Scheme 3),20,23 the stereochemical outcome of the reaction of enynes1 with cyclic alkenes was the opposite (Scheme 2).19 This stereochemical disparity led us to further explore reactions of enynes with a variety of cyclic and acyclic alkenes.
Herein we present experimental and theoretical evidence on the mechanism of the gold(I)-catalyzed intermolecular cyclopropanation of alkenes with enynes. Our results are consistent with a cyclopropanation taking place by the concerted or stepwise formation of two C–C bonds depending on the nature of the alkene. However, even in the stepwise case, the overall process takes place stereospecifically.
Results and discussion
Cyclopropanation of cyclic alkenes
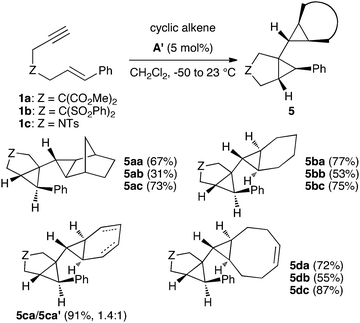
The cyclopropanation proceeded satisfactorily for catalystA′ with norbornene, cyclohexene, and 1,5-cyclooctadiene to give anti-adducts (Scheme 5). In this last case, the cycloaddition led cleanly to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adducts 5da–5dc as single stereoisomers. As expected, a mixture of diastereomers 5ca and 5ca′ was obtained in the reaction of 1,3-cyclohexadiene. The structures of biscyclopropanes 5ab, 5bc, and 5db were confirmed by X-ray diffraction (Fig. 2).
:1 adducts 5da–5dc as single stereoisomers. As expected, a mixture of diastereomers 5ca and 5ca′ was obtained in the reaction of 1,3-cyclohexadiene. The structures of biscyclopropanes 5ab, 5bc, and 5db were confirmed by X-ray diffraction (Fig. 2).
 | ||
| Fig. 2 X-ray structures of 5ab, 5bc, and 5db. | ||
Cyclopropanation of acyclic alkenes
The cyclopropanation of a series of substituted styrenes with 1,6-enyne 1c using different cationic gold(I) catalysts led to the corresponding cyclopropanes in good to excellent yields (Table 1).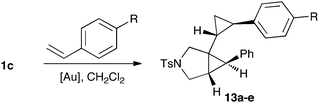
| Entry | R | [Au] | Product (yield, %) | dr |
|---|---|---|---|---|
| a Reactions at −40 °C and slowly warmed to r. t. (15 h), using 5 equiv. of the styrene and 5 mol% of catalyst. | ||||
| 1 | H | A | 13a (97) |
87:12:1:0 |
| 2 | H | B | 13a (83) |
61:36:3:0 |
| 3 | H | D | 13a (95) |
86:12:2:0 |
| 4 | MeO | A | 13b (80) |
75:23:2:0 |
| 5 | MeO | B | 13b (84) | 22:60:15:3 |
| 6 | MeO | D | 13b (70) |
73:23:4:0 |
| 7 | Cl | A | 13c (85) |
92:8:0:0 |
| 8 | Cl | D | 13c (90) |
89:10:1:0 |
| 9 | CF3 | A | 13d (98) |
93:7:0:0 |
| 10 | CF3 | D | 13d (82) |
92:7:1:0 |
| 11 | AcO | A | 13e (86) |
89:11:0:0 |
| 12 | AcO | D | 13e (93) |
84:14:2:0 |
A competitive study on the cyclopropanation of p-substituted styrenes with 1,6-enyne 1c using gold(I)-catalysts A and D showed good correlations with the σ Hammett constants (Fig. 3). As expected, the value of the slope ρ (−1.32 for A and −1.93 for D) shows that this cyclopropanation reaction is an electrophilic process. These results also confirm that the gold(I) intermediates 3 with L = phosphine are more electrophilic than those with more electron-donating L = N-heterocyclic carbene.11b
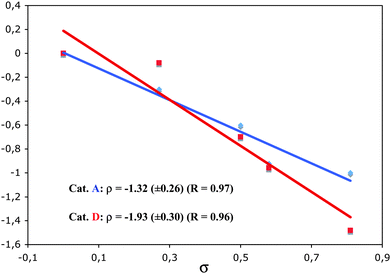 | ||
| Fig. 3 Hammett plots of relative rates for the cyclopropanation of styrenes with 1,6-enynes 1c using catalystsA (blue) and D (red). | ||
These results are consistent with those obtained in the cyclopropanation reaction catalyzed with other transition metals (Table 2).32,33,34 Thus, a ρ value of −2.4 was determined in the cyclopropanation of styrenes with the Simmons–Smith reagent,35 more negative than the value (−1.61) found for the reaction with ZnEt2 and CH2I2,36 or the one (−0.62) corresponding to the reaction with dichlorocarbene generated from phenyl(bromodichloromethyl)mercury.37 Studies on the reaction of styrenes with methyl phenyldiazoacetate showed a closer correlation with σ+ (ρ+ −1.0).38 In the cyclopropanation of styrenes with a series of dihydridobis(pyrazolyl)borate)copper complexes values of ρ ranging from −0.89 to −1.23 were obtained,39 whereas the reaction catalyzed by a bis(oxazoline)copper complex shows a better Hammett correlation with σ+ (ρ+ −0.51).40 Similar values have been obtained in other cyclopropanations proceeding through a carbene-transfer mechanism.41
The cyclopropanation can afford up to four different diastereomeric cyclopropanes; using catalystsA and D good stereoselectivities were observed (Table 1, entries 1, 3, 4, 7, 9 and 11). The configuration of the major stereoisomer 13a was assigned by X-ray crystallography (Fig. 4). Interestingly, the stereoselectivity decreased significantly in the reaction between 1c and styrene using IPr–Au(I) catalystB (Table 1, entry 2) and was reversed in the case of p-methoxystyrene (Table 1, entry 5).
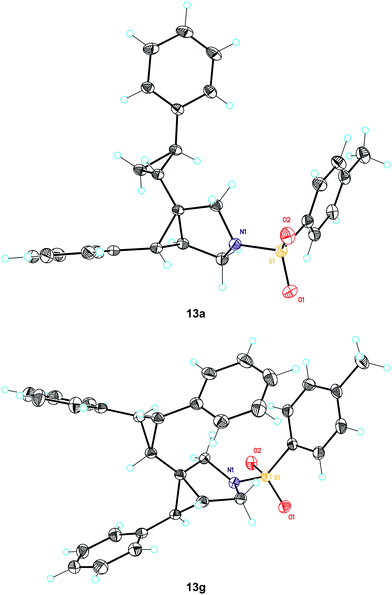 | ||
| Fig. 4 X-ray structures 13a and 13g. | ||
The gold(I)-catalyzed reaction of 1a with styrene gave a mixture of cyclopropanation products, from which the major stereoisomer 13f was isolated in 76% yield (Fig. 5). Similarly, reaction of 1c with trans-stilbene gave 13g in almost quantitative yield, whose configuration was confirmed by X-ray diffraction (Fig. 4). In sharp contrast, no reaction was observed with cis-stilbene. Indeed, a reaction of 1c with a 1:1 mixture of cis- and trans-stilbene led only to the recovery of unchanged starting materials. We also noted that no reaction took place between pivalate 8b (see Scheme 3) and cis-stilbene with gold complex D. Other gold(I)-catalyzed reactions were inhibited in the presence of cis-stilbene, which suggests that this alkene binds tightly to certain gold(I) complexes. 2-Methylpropene, 4-methylpenta-1,3-diene, and 2,5-dimethylhexa-2,4-diene provided 13h-j as the major stereoisomers. Interestingly, reaction of 1c with 2-allyl-1,3-diphenylpropane-1,3-dione led cleanly to the product of cyclopropanation 13k, whereas other 1,3-dicarbonyl compounds react with 1,6-enynes as C-nucleophiles.11b
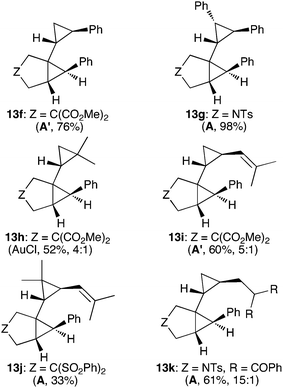 | ||
| Fig. 5 Products of cyclopropanation of acyclic alkenes. (The yield of 13j is based on a 70% conversion). | ||
The issue of the stereospecificity in the cyclopropanation was addressed using trans- and cis-β-methylstyrene as the alkenes (Scheme 6). In the first case, four diastereomers were observed, while with cis-β-methylstyrene, three cyclopropanes were obtained. Importantly, the reaction is stereospecific regarding the alkene as demonstrated by careful analysis of the 1H NMR spectra (Fig. 6), in keeping with that reported in gold(I)-catalyzed cyclopropanation of alkenes with propargyl carboxylates20,23 and other related metal-catalyzed processes.21Chromatography allowed for the separation of minor 13l33 and major 13m11 isomers, whose structures were assigned by X-ray diffraction (Fig. 7).
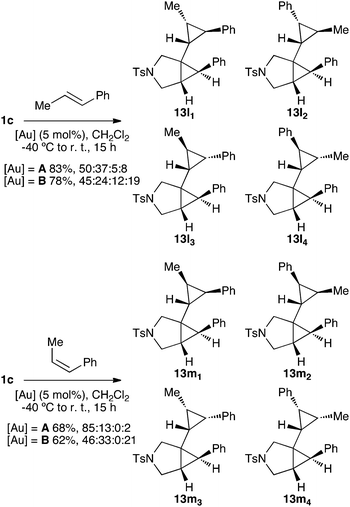 | ||
| Scheme 6 Products of cyclopropanation of (E)- and (Z)-β-methylstyrene with enyne1c. | ||
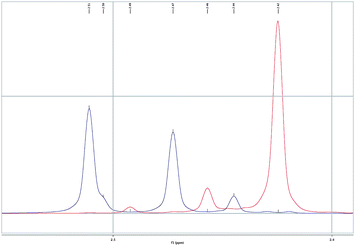 | ||
| Fig. 6 Section of the 1H NMR (CDCl3) spectra of the mixture of cyclopropyl derivatives 13l1–41–4 (blue) and 13m1–31–3 (red) showing no superposition of the N-methyl signals. | ||
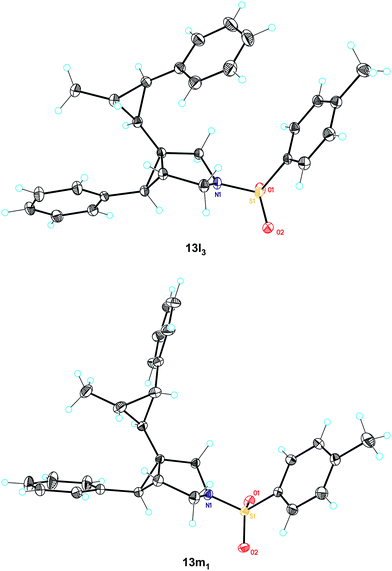 | ||
| Fig. 7 X-ray structures of 13l33 and 13m11. | ||
Mechanism and stereoselectivity
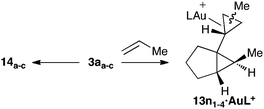
We also theoretically studied (DFT, B3LYP, 6-31G(d) (C, P, N, H) and SDD (Au))† the cyclopropanation of propene by intermediate 3a, which could lead to four diastereomeric cyclopropanes13n11–13n44 (Scheme 7). The lower activation energies were found for the cyclopropanation leading to diastereomers 13n1a–c1a–c (Table 3, entry 1 and Fig. 8), which is consistent with that found experimentally. Changing the ligand on the gold catalyst does not affect the observed trend, as the same preferred diastereomer was obtained when a simplified version of a N-heterocylic carbene, a phosphine (PMe3), and a phosphite (P(OMe)3) were considered. Since intermediate 3a could also evolve by single-cleavage exo-rearrangement to give cation14,6 we also computed this competitive intramolecular reaction at the same level of theory (Table 3, entry 5). The activation energies in this rearrangement are slightly higher than the ones found for the most favorable cyclopropanation pathway (0.8–2.3 kcal mol−1).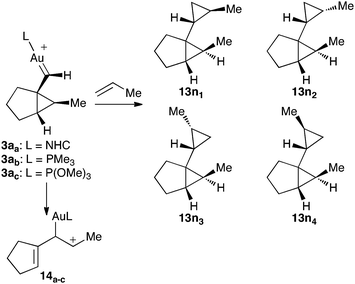 | ||
| Scheme 7 | ||
| Entry | TS | L = NHCb | L = PMe3 | L = P(OMe)3 |
|---|---|---|---|---|
| a Activation energies (ZPE-corrected potential energy in CH2Cl2) in kcal mol−1. b NHC = N,N′-dimethylimidazol-2-ylidene. | ||||
| 1 | TS(13n1a–c1a–c) | 7.5 | 7.9 | 8.4 |
| 2 | TS(13n2a–c2a–c) | 8.8 | 9.5 | 9.7 |
| 3 | TS(13n3a-c3a-c) | 11.4 | 11.3 | 11.2 |
| 4 | TS(13n4a-c4a-c) | 11.5 | 11.9 | 11.7 |
| 5 | TS(14) | 9.7 | 10.2 | 9.3 |
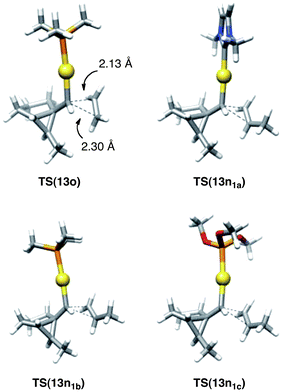 | ||
| Fig. 8 Comparison between the transition states for the reaction of 3abb with ethylene and those of 3aa–ca–c with propene. | ||
The cyclopropanation reaction of propene with 3aa–ca–c is a concerted reaction that proceeds through a moderately asynchronous transition state, in which the formation of the carbene–C1 bond is slightly more advanced (2.1–2.2 Å) than that with C2 (2.2–2.5 Å) (Fig. 8). A similar transition state TS(13o) was found for the reaction between 3abb (L = PMe3) and ethylene (activation energy = 10.2 kcal mol−1).
The products obtained in the two most favorable pathways leading to biscyclopropanes 13n11 and 13n22 can be described as corner-metalated cyclopropanes (average distances = 2.7 and 2.3 Å, respectively), while AuL+ is coordinated to one of the newly formed bonds (edge-coordinated) of the cyclopropane in 13n33 and 13n44 (average distances = 2.5 Å) (Fig. 9). A corner-metalated species was already found to be the primary product in the intramolecular gold-catalyzed cyclopropanation of dienynes.14
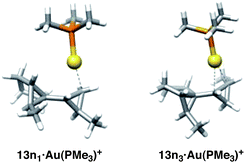 | ||
| Fig. 9 Corner- and edge-aurated products. | ||
In the cyclopropanation of styrene with 3b (L = PMe3), the most favorable pathway leads to stereosiomer 13p11 with the same relative configuration than 13n11 (Table 4), in agreement with that found experimentally.
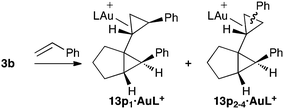
| Entry | TS | L = PMe3 |
|---|---|---|
| a Activation energies (ZPE-corrected potential energy in CH2Cl2) in kcal mol−1. | ||
| 1 | TS(13p11) | 8.7 |
| 2 | TS(13p22) | 11.3 |
| 3 | TS(13p33) | 11.9 |
| 4 | TS(13p44) | 11.7 |
However, in this case, formation of 13p11 was found to proceed stepwise via open intermediate 15, although its closure to form 13p11·AuL++ proceeds through TS(13p11)22 with a much lower barrier than that required for rotation around the PhCH–CH2 bond (C2–C3) (Fig. 10). As a consequence, although formally stepwise, the two C–C bonds in this cyclopropanation are also formed on the same side of the alkene.
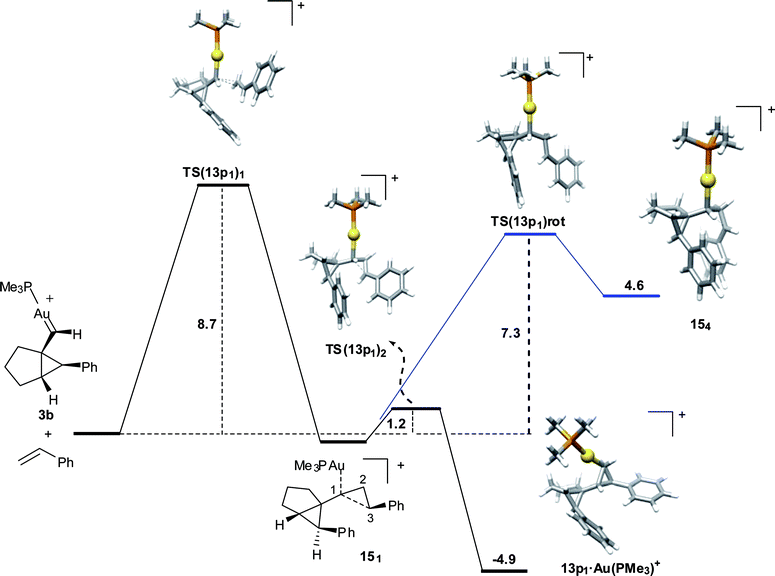 | ||
| Fig. 10 Reaction coordinate for the cyclopropanation of styrene with 3b leading to the major diastereomer 13p11. Activation energies (ZPE-corrected potential energy in CH2Cl2) in kcal mol−1 | ||
Intermediate 1511 (see Fig. 10 for atom labelling) presents a C1–C2–C3 angle of 95.3°, having one of the bonds (C1–C2) in the cyclopropane already formed (1.61 Å). The C1–C3 bond is still very elongated (2.26 Å) and will be completely formed through TS(13p11)22.
The reaction profile was additionally computed for trans-stilbene. Although gas-phase calculations led to a profile very similar to the one found for styrene in Fig. 10, when solvent corrections were applied, a concerted mechanism similar to that found for ethylene and propene was revealed.
The most favorable transition state TS(13p11) for the cyclopropanation of styrene with 3b is compared in Fig. 11 with that computed previously for the related intramolecular processes (TS(intramol)).14 The newly formed bond distances in TS(intramol) (2.23 and 2.10 Å) are shorter than the ones found in TS(13n11) (2.46 and 2.15 Å) for the cyclopropanation of propene with 3a. As expected, TS(13p11) is more distorted having bond lengths of 2.74 and 2.32 Å. In all cases, as expected for an electrophilic cyclopropanation, the longest distance corresponding to that with the substituted carbon in the alkene.
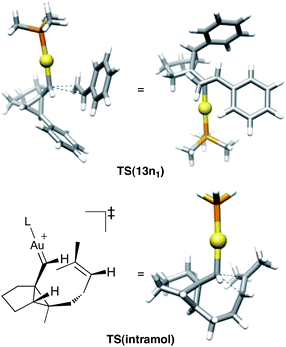 | ||
| Fig. 11 Comparison of the transition states for the gold(I)-catalyzed intermolecular cyclopropanation of styrene with an intramolecular process. | ||
In contrast to the gold(I)-catalyzed intermolecular cyclopropanation of alkenes with propargyl carboxylates,20,23 in the intermolecular cyclopropanation of alkenes with enynes the regio- and stereochemistry are mainly controlled by the substituent at C1 of the alkene (Fig. 12). According to these models, increasing the steric hindrance of the ligand should result in a deterioration of the stereoselectivity, favoring new transitions states TS(13n2a–c2a–c) and TS(13p22). This is in agreement with the experimental results shown in Table 1 (entries 4 and 5) in the reaction between enyne1c and p-methoxystyrene changing from catalystA to B (from IMes to IPr ligand). This model also explains the regiochemistry observed for 4-methylpenta-1,3-diene (Fig. 5), in which the cyclopropanation occurs at the less sterically hindered alkene to form 13i.
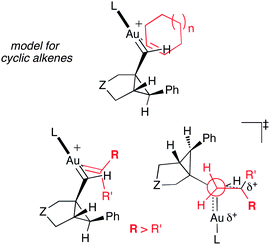 | ||
| Fig. 12 Models for the gold(I)-catalyzed intermolecular cyclopropanation of cyclic and acyclic alkenes. | ||
Conclusions
Our work shows that the intermediates in gold(I)-catalyzed cyclizations of enynes are not simple carbocations. These intermediates react with alkenes to give cyclopropanes in a stereospecific process in which the configuration of the alkene is preserved, which is consistent with that found in the gold(I)-catalyzed cyclopropanation of alkenes with propargyl carboxylates.20,23 Our results complement a recent theoretical study,30 which showed that reactions with highly polarized alkenes such as methyl vinyl ether take place in a stepwise fashion. From our results, we conclude that the cyclopropanation can occur both in a concerted or stepwise fashion depending on the substituents present in the alkene. Thus, whereas the reaction with ethylene and propene is concerted, the cyclopropanation with the more polarized styrene is stepwise. However, even in this case, the stereochemical outcome of the reaction is the same: the two new C–C bonds are formed from the same face of the alkene.The gold(I)-catalyzed cyclopropanation of alkenes with 1,6-enynes is an electrophilic process, related to other well established cyclopropanation reactions that proceed via metal carbenes or carbenoids such as the Simmons–Smith reaction. Although highly reactive, cyclopropylgoldcarbenes formed in cyclizations of 1,6-enynes are less electrophilic than the zinc carbenoids of the Simmons–Smith cyclopropanation.
Acknowledgements
We thank the MICINN (CTQ2010-16088/BQU, CTQ2008-06866-C02-02, Consolider Ingenio 2010 Grant CSD2006-0003, and FPI predoctoral fellowship to P.P.-G.), the MEC (FPU predoctoral fellowship to E.H.-G.), the AGAUR (2009 SGR 47, 2009 SGR 259), and the ICIQ Foundation for support of this work.Notes and references
- (a) E. Jiménez-Núñez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326–3350 CrossRef CAS; (b) D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351–3378 CrossRef CAS; (c) V. Michelet, P. Y. Toullec and J.-P. Genêt, Angew. Chem., Int. Ed., 2008, 47, 4268–4315 CrossRef CAS; (d) A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180–3211 CrossRef; (e) A. Fürstner, Chem. Soc. Rev., 2009, 38, 3208–3221 RSC.
- L. Zhang, J. Sun and S. A. Kozmin, Adv. Synth. Catal., 2006, 348, 2271–2296 CrossRef CAS.
- (a) C. Nieto-Oberhuber, M. P. Muñoz, E. Buñuel, C. Nevado, D. J. Cárdenas and A. M. Echavarren, Angew. Chem., Int. Ed., 2004, 43, 2402–2406 CrossRef CAS; (b) C. Nieto-Oberhuber, M. P. Muñoz, S. López, E. Jiménez-Núñez, C. Nevado, E. Herrero-Gómez, M. Raducan and A. M. Echavarren, Chem.–Eur. J., 2006, 12, 1677–1693 CrossRef CAS.
- C. Nieto-Oberhuber, S. López, E. Jiménez-Núñez and A. M. Echavarren, Chem.–Eur. J., 2006, 12, 5916–5923 CrossRef CAS.
- S. I. Lee and N. Chatani, Chem. Commun., 2009, 371–384 RSC.
- C. Nieto-Oberhuber, S. López, M. P. Muñoz, D. J. Cárdenas, E. Buñuel, C. Nevado and A. M. Echavarren, Angew. Chem., Int. Ed., 2005, 44, 6146–6148 CrossRef CAS.
- C. Nevado, D. J. Cárdenas and A. M. Echavarren, Chem.–Eur. J., 2003, 9, 2627–2635 CrossRef CAS.
- (a) L. Zhang and S. A. Kozmin, J. Am. Chem. Soc., 2005, 127, 6962–6963 CrossRef CAS; (b) A. K. Buzas, F. M. Istrate and F. Gagosz, Angew. Chem., Int. Ed., 2007, 46, 1141–1144 CrossRef CAS; (c) B. D. Sherry, L. Maus, B. N. Laforteza and F. D. Toste, J. Am. Chem. Soc., 2006, 128, 8132–8133 CrossRef CAS; (d) S. Park and D. Lee, J. Am. Chem. Soc., 2006, 128, 10664–10665 CrossRef CAS; (e) Y. Horino, M. R. Luzung and F. D. Toste, J. Am. Chem. Soc., 2006, 128, 11364–11365 CrossRef CAS; (f) C. A. Witham, P. Mauleón, N. D. Shapiro, B. D. Sherry and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 5838–5839 CrossRef CAS; (g) P. Y. Toullec, T. Blarre and V. Michelet, Org. Lett., 2009, 11, 2888–2891 CrossRef CAS; (h) S. G. Sethofer, T. Mayer and F. D. Toste, J. Am. Chem. Soc., 2010, 132, 8276–8277 CrossRef CAS.
- A. Fürstner and L. Morency, Angew. Chem., Int. Ed., 2008, 47, 5030–5033 CrossRef.
- (a) C. Nieto-Oberhuber, S. López and A. M. Echavarren, J. Am. Chem. Soc., 2005, 127, 6178–6179 CrossRef CAS; (b) C. Nieto-Oberhuber, P. Pérez-Galán, E. Herrero-Gómez, T. Lauterbach, C. Rodríguez, S. López, C. Bour, A. Rosellón, D. J. Cárdenas and A. M. Echavarren, J. Am. Chem. Soc., 2008, 130, 269–279 CrossRef CAS.
- (a) C. H. M. Amijs, C. Ferrer and A. M. Echavarren, Chem. Commun., 2007, 698–700 RSC; (b) C. H. M. Amijs, V. López-Carrillo, M. Raducan, P. Pérez-Galán, C. Ferrer and A. M. Echavarren, J. Org. Chem., 2008, 73, 7721–7730 CrossRef CAS.
- P. Y. Toullec, E. Genin, L. Leseurre, J.-P. Genêt and V. Michelet, Angew. Chem., Int. Ed., 2006, 45, 7427–7430 CrossRef CAS.
- (a) B. M. Trost and A. S. K. Hashmi, Angew. Chem., Int. Ed., 1993, 32, 1085–1087 CrossRef; (b) B. M. Trost and A. S. K. Hashmi, J. Am. Chem. Soc., 1994, 116, 2183–2184 CrossRef CAS; (c) B. M. Trost, A. S. K. Hashmi and R. G. Ball, Adv. Synth. Catal., 2001, 343, 490–494 CrossRef CAS.
- C. Nieto-Oberhuber, S. López, M. P. Muñoz, E. Jiménez-Núñez, E. Buñuel, D. Cárdenas and A. M. Echavarren, Chem.–Eur. J., 2006, 12, 1694–1702 CrossRef CAS.
- S. M. Kim, J. H. Park, S. Y. Choi and Y. K. Chung, Angew. Chem., Int. Ed., 2007, 46, 6172–6175 CrossRef CAS.
- N. Chatani, K. Kataoka, S. Murai, N. Furukawa and Y. Seki, J. Am. Chem. Soc., 1998, 120, 9104–9105 CrossRef CAS.
- (a) E. Mainetti, V. Mouriès, L. Fensterbank, M. Malacria and J. Marco-Contelles, Angew. Chem., Int. Ed., 2002, 41, 2132–2135 CrossRef CAS; (b) Y. Harrak, C. Blaszykowski, M. Bernard, K. Cariou, E. Mainetti, V. Mouriès, A.-L. Dhimane, L. Fensterbank and M. Malacria, J. Am. Chem. Soc., 2004, 126, 8656–8657 CrossRef CAS.
- B. P. Peppers and S. T. Diver, J. Am. Chem. Soc., 2004, 126, 9524–9525 CrossRef CAS.
- S. López, E. Herrero-Gómez, P. Pérez-Galán, C. Nieto-Oberhuber and A. M. Echavarren, Angew. Chem., Int. Ed., 2006, 45, 6029–6032 CrossRef CAS.
- M. J. Johansson, D. J. Gorin, S. T. Staben and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 18002–18003 CrossRef CAS.
- See also: (a) K. Miki, K. Ohe and S. Uemura, Tetrahedron Lett., 2003, 44, 2019–2022 CrossRef CAS; (b) K. Miki, K. Ohe and S. Uemura, J. Org. Chem., 2003, 68, 8505–8513 CrossRef CAS; (c) K. Miki, T. Yokoi, F. Nishino, Y. Kato, Y. Washitake, K. Ohe and S. Uemura, J. Org. Chem., 2004, 69, 1557–1564 CrossRef CAS; (d) K. Miki, F. Nishino, K. Ohe and S. Uemura, J. Am. Chem. Soc., 2002, 124, 5260–5261 CrossRef.
- Cyclopropanation by gold(I)-catalyzed reaction of diazoacetates: (a) M. R. Fructos, T. R. Belderrain, P. de Frémont, N. M. Scott, S. P. Nolan, M. M. Díaz-Requejo and P. J. Pérez, Angew. Chem., Int. Ed., 2005, 44, 5284–5288 CrossRef CAS; (b) A. Prieto, M. R. Fructos, M. M. Díaz-Requejo, P. J. Pérez, P. Pérez-Galán, N. Delpont and A. M. Echavarren, Tetrahedron, 2009, 65, 1790–1793 CrossRef CAS.
- D. Benitez, N. D. Shapiro, E. Tkatchouk, Y. Wang, W. A. Goddard III and F. D. Toste, Nat. Chem., 2009, 1, 482–486 CrossRef CAS.
- (a) A. Fedorov, M.-E. Moret and P. Chen, J. Am. Chem. Soc., 2008, 130, 8880–8881 CrossRef CAS; (b) A. Fedorov and P. Chen, Organometallics, 2009, 28, 1278–1281 CrossRef CAS.
- (a) E. Herrero-Gómez, C. Nieto-Oberhuber, S. López, J. Benet-Buchholz and A. M. Echavarren, Angew. Chem., Int. Ed., 2006, 45, 5455–5459 CrossRef CAS; (b) P. Pérez-Galán, N. Delpont, E. Herrero-Gómez, F. Maseras and A. M. Echavarren, Chem.–Eur. J., 2010, 16, 5324–5332 CAS.
- C. Bartolomé, Z. Ramiro, D. García-Cuadrado, P. Pérez-Galán, M. Raducan, C. Bour, A. M. Echavarren and P. Espinet, Organometallics, 2010, 29, 951–956 CrossRef CAS.
- G. Seidel, R. Mynott and A. Fürstner, Angew. Chem., Int. Ed., 2009, 48, 2510–2513 CrossRef CAS.
- See also: (a) A. S. K. Hashmi, Angew. Chem., Int. Ed., 2008, 47, 6754–6756 CrossRef CAS; (b) C.-M. Chao, M. R. Vitale, P. Y. Toullec, J.-P. Genêt and V. Michelet, Chem.–Eur. J., 2009, 15, 1319–1323 CrossRef CAS.
- D. L. Boger and C. E. Brotherton, Tetrahedron Lett., 1984, 25, 5611–5614 CrossRef CAS.
- L. Batiste, A. Fedorov and P. Chen, Chem. Commun., 2010, 46, 3899–3901 RSC.
- E. Jiménez-Núñez, C. K. Claverie, C. Bour, D. J. Cárdenas and A. M. Echavarren, Angew. Chem., Int. Ed., 2008, 47, 7892–7895 CrossRef CAS.
- H. M. L. Davies and E. G. Antoulinakis, Org. React., 2001, 57, 1–326 CAS.
- (a) M. P. Doyle, Acc. Chem. Res., 1986, 19, 348–356 CrossRef CAS; (b) M. P. Doyle, J. H. Griffin, V. Bagheri and R. L. Dorow, Organometallics, 1984, 3, 53–61 CrossRef CAS.
- Lead general references: (a) H. Lebel, J.-F. Marcoux, C. Molinaro and A. B. Charette, Chem. Rev., 2003, 103, 977–1050 CrossRef CAS; (b) W. A. Donaldson, Tetrahedron, 2001, 57, 8589–8627 CrossRef CAS.
- N. Kawabata, I. Kamemura and M. Naka, J. Am. Chem. Soc., 1979, 101, 2139–2145 CrossRef CAS.
- J. Nishimura, J. Furukawa, N. Kawabata and M. Kitayama, Tetrahedron, 1971, 27, 1799–1806 CrossRef.
- D. Seyferth, J. Y.-P. Mui and R. Damrauer, J. Am. Chem. Soc., 1968, 90, 6182–6186 CrossRef CAS.
- H. M. L. Davies and S. A. Panaro, Tetrahedron, 2000, 56, 4871–4880 CrossRef CAS.
- (a) M. M. Díaz-Requejo, P. J. Pérez, M. Brookhart and J. L. Templeton, Organometallics, 1997, 16, 4399–4402 CrossRef CAS; (b) M. M. Díaz-Requejo, M. C. Nicasio and P. J. Pérez, Organometallics, 1998, 17, 3051–3057 CrossRef CAS.
- T. Rasmussen, J. F. Jensen, N. Østergaard, D. Tanner, T. Ziegler and P.-O. Norrby, Chem.–Eur. J., 2002, 8, 177–184 CrossRef CAS.
- P. Müller, Acc. Chem. Res., 2004, 37, 243–251 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental results and computational data. X-ray crystallographic data: CCDC 780815–780821. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00335b |
| ‡ Additional address: Departament de Química, Universitat Autónoma de Barcelona, 08193 Bellaterra, Spain |
| § Additional address: Departament de Química Analítica i Química Orgànica, Universitat Rovira i Virgili, C/Marcel·li Domingo s/n, 43007 Tarragona, Spain |
| This journal is © The Royal Society of Chemistry 2011 |