Donor-substituted octacyano[4]dendralenes: a new class of cyano-rich non-planar organic acceptors†
Benjamin
Breiten
a,
Yi-Lin
Wu
a,
Peter D.
Jarowski
a,
Jean-Paul
Gisselbrecht
b,
Corinne
Boudon
b,
Markus
Griesser
c,
Christine
Onitsch
c,
Georg
Gescheidt
c,
W. Bernd
Schweizer
a,
Nicolle
Langer
d,
Christian
Lennartz
e and
François
Diederich
*a
aLaboratorium für Organische Chemie, ETH Zürich, Hönggerberg, HCI, CH-8093, Zürich, Switzerland. E-mail: diederich@org.chem.ethz.ch; Fax: (+41)-44-632-1109
bLaboratoire d'Electrochimie et de Chimie Physique du Corps Solide, Institut de Chimie - UMR 7177, Université de Strasbourg, C.N.R.S., 4, rue Blaise Pascal, 67000, Strasbourg, France
cInstitute of Physical and Theoretical Chemistry, Graz University of Technology, Technikerstrasse 4/I, A-8010, Graz, Austria
dBASF SE, GVP/C-A30, 67056, Ludwigshafen, Germany
eBASF SE, GVC/E-B9, 67056, Ludwigshafen, Germany
First published on 3rd November 2010
Abstract
Double [2+2] cycloaddition/retro-electrocyclisation reactions between tetracyanoethene (TCNE) and various anilino-capped buta-1,3-diynes furnished a series of octacyano[4]dendralene derivatives featuring intense, low-energy intramolecular charge-transfer absorptions. These novel chromophores are strong electron acceptors and undergo facile one-electron reductions at potentials (–0.09 to –0.17 eV vs.Fc+/Fc, in CH2Cl2–0.1 M nBu4NPF6) lower than those reported for the benchmark organic acceptors, such as TCNE (–0.32 eV) and 7,7,8,8-tetracyanoquinodimethane (TCNQ) (–0.25 eV). The electron-accepting power of one octacyano[4]dendralene, as expressed by the computed adiabatic electron affinity (EA), compares to that of the reference acceptor 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4-TCNQ) used as a p-type dopant in organic light-emitting diodes (OLEDs) and solar cells. Gas-phase density functional theory (DFT) calculations predict a stretched-out conformation as the global energy minimum for octacyano[4]dendralenes. In the solid state however, folded conformations were observed for two structures by X-ray analysis. Taking the solid state environment approximately into account calculations predict a energetical degeneracy between the stretched-out and folded conformation. Therefore conformational preference probably is a result of supramolecular dimer formation, mediated by two pairs of intermolecular, antiparallel dipolar CN⋯CN interactions.
Introduction
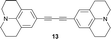
Among the large number of organic electron-acceptors, cyano-rich representatives are the most prominent class of compounds for opto-electronic devices,1 due to the strong accepting power compared to the small molecular weight of the cyano group. Tetracyanoethene (TCNE),2,37,7,8,8-tetracyanoquinodimethane (TCNQ)4,5 and 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4-TCNQ), together with their derivatives, have found widespread application in organic light-emitting diodes (OLEDs), organic field-effect transistors (OFETs) and organic solar cells.6 Furthermore, environmentally stable acceptors are increasingly explored as p-type dopants, significantly improving the performance of opto-electronic devices. Although F4-TCNQ is widely used for this purpose, alternative dopants with superior thermal stability are currently being pursued. Recently, the [2+2] cycloaddition of TCNE, TCNQ and F4-TCNQ to electron-rich alkynes, followed by retro-electrocyclisation,7,8 has provided a large number of non-planar push–pull chromophores featuring 1,1,4,4-tetracyano- and 1,1,2,4,4-pentacyanobuta-1,3-dienes as well as cyclohexa-2,5-diene-1,4-diylidene-expanded derivatives as electron acceptor components.9 They show intense, bathochromically shifted intramolecular charge-transfer bands and, despite substitution by strong donors such as anilines, ferrocene or tetrathiafulvene (TTF), compete with TCNE, TCNQ and F4-TCNQ in their ease of reversible electron uptake. Some of them display high third-order optical nonlinearities and have in the meantime been applied to the development of opto-electronic devices, such as waveguides.10 Based on these results, we expected that even stronger electron-acceptors would be in reach upon increasing the number of electron-withdrawing cyano functionalities in the molecule.While we had earlier taken advantage of the fact that so-called “electronically confused” acetylenes with an anilino donor and a cyano acceptor substituent also undergo this transformation,11 we had not achieved the sequential double addition of TCNE to donor-activated buta-1,3-diynes under the formation of novel chromophores featuring octacyano[4]dendralene12,13 structures as electron acceptors. Based on IR-spectroscopic evidence, such a structure had been proposed by Bruce et al. as the product of a double TCNE addition to a bis[Ru(PPh3)2Cp]-end-capped octa-1,3,5,7-tetrayne.14 Here, we report the synthesis of the donor-substituted octacyano[4]dendralenes 1–7 by double addition of TCNE to buta-1,3-diynes 8–14, activated by two anilino donors, as well as the fascinating structural and physical properties of the new push–pull chromophores.
Results and discussion
Synthesis
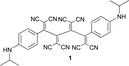
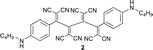
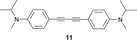
Mono-addition of TCNE to bis(dimethylanilino)-end-capped butadiyne 10 occurs in nearly quantitative yield (isolated: 96%) by stirring equimolar amounts of the starting materials in solvents such as benzene, dichloromethane, dimethylformamide (DMF) or tetrahydrofuran (THF) at 20 °C for up to 1 h.8,15 The product, 2-[4-(dimethylamino)phenyl]-3-([4-(dimethylamino)phenyl]ethynyl)buta-1,3-diene-1,1,4,4-tetracarbonitrile (“DDMEBT”)10 does not produce any bis-adduct with an excess of TCNE at 20 °C, even at prolonged reaction times. At 95 °C in 1,1,2,2-tetrachloroethane however, bis-TCNE addition to 10 occurred and provided octacyano[4]dendralene 3 in 80% yield, after stirring for 4 d (Table 1).
| Entry | Buta-1,3-diyne | Product | Yield (%) |
|---|---|---|---|
| 1 |

|
|
82 |
| 2 |
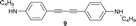
|
|
79 |
| 3 |

|
|
79 |
| 4 |
|
|
81 |
| 5 |

|
|
84 |
| 6 |
|
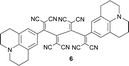
|
80 |
| 7 |

|
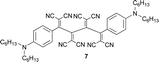
|
87 |
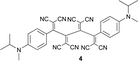
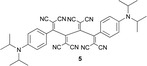
Anilines bearing larger, better solubilising N-alkyl groups undergo a more rapid conversion. Thus, N-monoalkylated bis-adducts 1 and 2 were obtained in 79–82% yield upon heating the corresponding diynes with TNCE (2 equiv.) at 80 °C for 2 d, whereas N,N-dialkylated 4–7 were prepared in 1 d in 79–87% yield (Table 1). It is noticeable that the bis-addition to these anilino-capped butadiynes with the larger, better solubilising N-alkyl substituents also occurs at 20 °C, although at a much slower rate.
The constitution of the donor-substituted octacyano[4]dendralenes was unambiguously assigned by X-ray analyses (vide infra). These new push–pull systems are highly coloured, stable solids that can be stored for months in laboratory atmosphere without decomposition and they feature extraordinary high melting points (>400 °C).
X-Ray crystallography
Single crystals of 5 and 7 were grown by slow diffusion of hexane into CH2Cl2 solutions of the compounds at 20 °C.‡ Analysis of the X-ray crystal structures unexpectedly revealed an octacyano[4]dendralene skeleton, in which all eight cyano groups converge into one hemisphere, whereas the two anilino rings undergo π–π stacking. The torsion angles about the central C–C bond (C2–C3–C4–C5) of the dendralene scaffold amount to ca. −51° for 5 and ca. −43° for 7 which leads to an overall rather unexpected syn-conformation of the molecules (Fig. 1).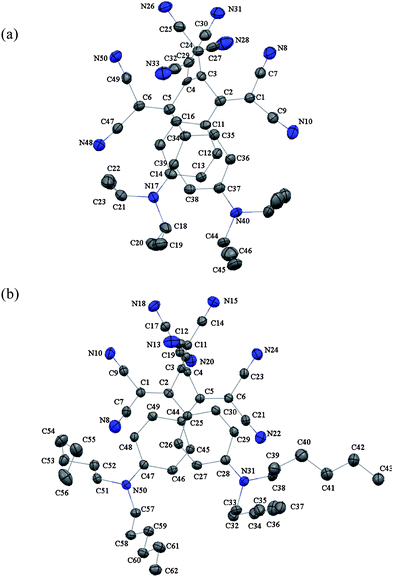 | ||
| Fig. 1 Molecular structures (ORTEP plots) of 5 (a) and 7 (b) (Mercury,16 ellipsoids drawn at 173 K for 5 and 123K for 7 at the 50% probability level. Arbitrary numbering. H-atoms are omitted.) | ||
While other interactions such as π-stacking are certainly relevant in determining the crystal packing, we propose that the intermolecular dipolar CN⋯CN interactions, revealed by the crystal structures, enforce the syn conformation (Fig. 2). Two neighbouring molecules interact closely with their strong CN dipoles,17 thereby undergoing favourable antiparallel dipolar interactions,18 involving two pairs of CN moieties in 5 and in 7 (Fig. 2; d(C⋯N) = 3.18, 3.55 Å for 5 and d(C⋯N) = 3.16, 3.55 Å for 7).
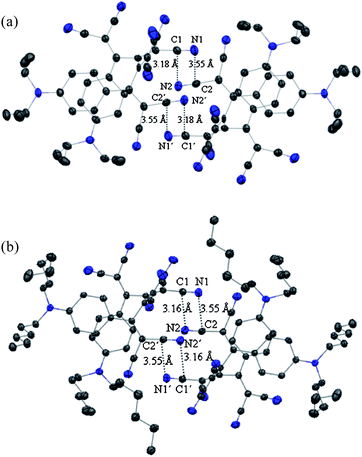 | ||
| Fig. 2 Arrangement of neighbouring molecules in the crystal packing showing short intermolecular contacts of 5 (a) and 7 (b) (Mercury,16 ellipsoids drawn at 173 K at the 50% probability level). | ||
Gas-phase calculations at the B3LYP/6-31G(d) level (see ESI† for details) predict a global minimum for compound 5 with a distinctly different conformation from the one found in the crystal. In these calculations, the extended anti-conformation is energetically preferred. The calculated global minima (extended geometry) for the N,N-dimethylanilino derivative 3 (a model for 7) and the N,N-diisopropylanilino derivative 5 are presented in Fig. 3 (right) along with the conformations (compact geometry) corresponding more closely to those found in the crystal structures (left).
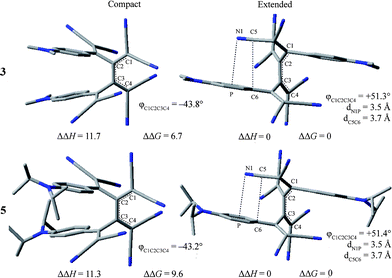 | ||
| Fig. 3 Optimised B3LYP/6-31G(d) gas-phase structures of compounds 3 and 5 in their compact (left) and extended (right) conformations. Relative enthalpies and free-energies are given (kJ mol−1). Highlighted structural parameters (° and Å) indicate the torsion angle about the central C2–C3 bonds (φC1C2C3C4) and distances between the nitrile and proximal N,N-dialkylanilino ring (dN1P, with P defined as the centre of the ring, and dC5C6) in the extended conformations. | ||
The minima adopt extended C2-symmetric conformations and the change in torsion angle about the central C–C single bonds of the [4]dendralene backbones primarily accounts for the difference between the gas-phase and solid-state structures. The calculated compact gas-phase structures, corresponding to the observed solid-state structures, are significantly higher in energy (for 3 (ΔΔH = 11.7 kJ mol−1, ΔΔG = 6.7 kJ mol−1) and 5 (ΔΔH = 11.3 kJ mol−1, ΔΔG = 9.6 kJ mol−1) than the calculated extended geometries. Using a slightly different computational approach two local minima were identified for compound 5 in gas-phase geometry optimisations at BP86/SV(P)19 level of theory as well. One conformation (torsion angle −34.9°) is closely related to the compact molecular structure seen in the X-ray crystal structure, whereas the conformation corresponding to the second local minimum exhibits a more extended molecular geometry (torsion angle 47.5°). The gas-phase energetics (B3LYP/TZVP//BP86/SV(P), Table 2) suggest in agreement with the B3LYP/6-31G(d) calculations above, that the extended structure with the torsion angle of 47.5° is the most stable conformation. By simulating the solid-state environment by a dielectric continuum (COSMO, conductor-like screening mode)19 at the same level of theory however, the two structures become nearly degenerate. These findings suggest, that the torsional mode of the central C–C bond is very soft in the solid-state environment so that the additional antiparallel dipolar CN⋯CN interactions between the monomers present in the crystal structure account most likely for the fact that the compact geometry is preferred.
| Conformer 5 | ΔE/kJ mol−1 | ΔE(COSMO, ε = 4.5)/kJ mol−1 | ΔE(COSMO, ε = 4.5) + ΔZPE/kJ mol−1 |
|---|---|---|---|
| Compact (–34.9°) | 10.47 | 0.0 | 0.0 |
| Extended (47.5°) | 0.0 | 1.26 | 0.2 |
Electronic absorption spectroscopy
The UV/Vis spectra of the chromophores recorded in CH2Cl2 display an intense intramolecular charge-transfer (CT) band with end-absorptions reaching into the near infrared (NIR) region (Fig. 4, Table S2, ESI†). With molar extinction coefficients up to ε = 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 dm3 mol−1 cm−1, this band results from intramolecular charge-transfer between strongly coupled, linearly π-conjugated proximal donor and acceptor moieties. The energy of the transition progressively decreases as the anilino donor strength increases. Thus, λmax appears at 475 nm (2.61 eV) in 1, at 494 nm (2.51 eV) in 3, at 506 nm (2.45 eV) in 5, and at 524 nm (2.37 eV) in 6. The electrochemical data for the corresponding dianilino-end-capped buta-1,3-diyne starting materials (Table S3, ESI†) clearly show that the ease of oxidation of the anilino moieties and therefore the donor strength increases in this series. In addition to this strong band, there is probably a second, very weak CT transition around 700 nm, leading to the tailing of the end-absorption into the NIR region.20a This low-energy CT transition involves weakly coupled donor and acceptor moieties; in other words, most probably a charge transfer from the aniline to the cross-conjugated second (distal) tetracyanobutadiene moiety at the centre of the molecule.20b
000 dm3 mol−1 cm−1, this band results from intramolecular charge-transfer between strongly coupled, linearly π-conjugated proximal donor and acceptor moieties. The energy of the transition progressively decreases as the anilino donor strength increases. Thus, λmax appears at 475 nm (2.61 eV) in 1, at 494 nm (2.51 eV) in 3, at 506 nm (2.45 eV) in 5, and at 524 nm (2.37 eV) in 6. The electrochemical data for the corresponding dianilino-end-capped buta-1,3-diyne starting materials (Table S3, ESI†) clearly show that the ease of oxidation of the anilino moieties and therefore the donor strength increases in this series. In addition to this strong band, there is probably a second, very weak CT transition around 700 nm, leading to the tailing of the end-absorption into the NIR region.20a This low-energy CT transition involves weakly coupled donor and acceptor moieties; in other words, most probably a charge transfer from the aniline to the cross-conjugated second (distal) tetracyanobutadiene moiety at the centre of the molecule.20b
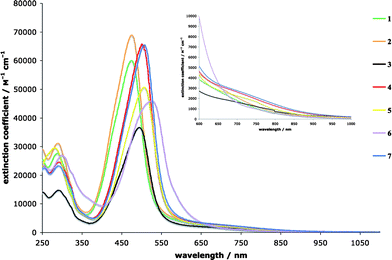 | ||
| Fig. 4 UV/Vis spectra of 1–7 in CH2Cl2 at 298 K. Inset: zoom on the low-energy CT bands. | ||
Electrochemistry
The redox properties of push–pull systems 1–7 were studied by cyclic voltammetry (CV) and rotating-disc voltammetry (RDV) in CH2Cl2 (+0.1 M nBu4NPF6, all potentials vs. the ferrocenium–ferrocene couple (Fc+/Fc)) and are summarised in Table 3. The two anilino donor moieties in chromophores1–7 undergo a reversible, two-electron oxidation step. From the peak shape characteristics, this electron transfer corresponds to an overlap of two reversible one-electron transfers occurring on the two non-interacting aniline moieties.| CV | RDV | |||
|---|---|---|---|---|
| E°/Va | ΔEp/mVb | E 1/2/Vc | Slope/mVd | |
| a E° = (Epc + Epa)/2, where Epc and Epa correspond to the cathodic and anodic peak potentials, respectively. b ΔEp = Eox − Ered, where the subscripts ox and red refer to the conjugated oxidation and reduction steps, respectively. c E p = Irreversible peak potential. d Slope = Slope of the linearised plot of E vs. log[I/(Ilim − I)], where Ilim is the limiting current and I the current. | ||||
| 1 | +1.04 | 80 | +1.02 (2e−) | 75 |
| –0.10 | 75 | –0.12 (1e−) | 70 | |
| –0.63 | 100 | –0.65 (1e−) | 95 | |
| –1.57 | 70 | –1.61 (1e−) | 60 | |
| –1.76 | 70 | –1.78 (1e−) | 70 | |
| 2 | +1.09 | 60 | ||
| +1.00 | 60 | +1.05 (2e−) | 100 | |
| –0.10 | 90 | –0.13 (1e−) | 75 | |
| –0.63 | 110 | –0.67 (1e−) | 70 | |
| –1.58 | 90 | |||
| −1.77 | 90 | |||
| 3 | +0.99 | 90 | +1.00 (2e−) | 70 |
| –0.09 | 85 | –0.10 (1e−) | 60 | |
| –0.60 | 100 | –0.67 (1e−) | 75 | |
| 4 | +0.98 | 90 | +0.98 (2e−) | 60 |
| –0.13 | 80 | –0.15 (1e−) | 70 | |
| –0.68 | 120 | –0.66 (1e−) | 90 | |
| –1.62 | 80 | –1.61 (1e−) | 60 | |
| –1.79 | 60 | –1.80 (1e−) | 80 | |
| 5 | +0.99 (2e−) | 70 | +0.98 (2e−) | 60 |
| –0.08 (1e−) | 70 | –0.09 (1e−) | 75 | |
| –0.58 (0.5e−) | 60 | –0.62 (1e−) | 85 | |
| –0.66 (0.5e−) | 60 | |||
| –1.56 (1e−) | 70 | –1.70 (2e−) | 140 | |
| –1.71 (1e−) | 70 | |||
| 6 | +0.77 | 80 | +0.79 (2e−) | 75 |
| –0.17 | 80 | –0.17 (1e−) | 60 | |
| –0.63 | 60 | –0.65 (0.5e−) | 60 | |
| –0.75 | 60 | –0.80 (0.5e−) | 60 | |
| –1.66 | 60 | –1.70 (1e−) | 60 | |
| –1.82 | 70 | –1.85 (1e−) | 70 | |
| 7 | +0.99 (2e−) | 80 | +1.00 (2e−) | 60 |
| –0.12 (1e−) | 90 | –0.11 (1e−) | 60 | |
| –0.61 (0.5e−) | 70 | –0.65 (1e−) | 90 | |
| –0.69 (0.5e−) | 70 | –1.69 (1e−) | 65 | |
| –1.63 (1e−) | 85 | –1.80 (1e−) | 90 | |
| –1.79 (1e−) | 75 | |||
More importantly, the studied compounds undergo four reversible one-electron reduction steps centred on the four dicyanovinyl moieties, except for 3, where only two one-electron steps are observed due to the insufficient solubility of the reduced species. Gratifyingly, despite the substitution with two potent anilino donors, the first reduction of 1–7 (–0.09 to −0.17 V) occurs at lower potentials than that of TCNE (–0.32 V) and TCNQ (–0.25 V) and the second electron uptake is also greatly facilitated (1–7: −0.65 to −0.80 V; TCNE: −1.35 V; TCNQ: −0.81 V).
By CV, the first reduction is perfectly reversible, whereas the peak characteristics and shape of the second reduction step did not correspond to a reversible one-electron transfer. Indeed, the peak potential difference (Epa − Epc) is much higher than the expected value of 58 mV for a reversible one-electron transfer and remains constant with scan rates. For 1–4, the second reduction also involves two overlapping electron transfers, unresolved for the latter species, explaining a larger peak potential difference. All these characteristics indicate an electron exchange per step of 0.5 e−. Such an unexpected behaviour may be explained for 5 by intense interactions between the electrogenerated two-electron reduced species and the one-electron reduced species obtained after the first reduction step forming a “dimer” which is reduced further to the “tetra-anionic dimer” as proposed in Scheme 1. The latter dissociates to the corresponding dianions. Such dimer formation under electrochemical control has been observed previously for porphyrins and corroles.21 It should be mentioned that in a more solvating medium, namely CH3CN, the second reduction step is not split anymore and has the characteristics of a reversible one-electron transfer.
 | ||
| Scheme 1 Proposed reduction scheme for compound 5, supported by the electrochemical data. | ||
The optical HOMO–LUMO gaps, determined from the end-absorption λend of the longest-wavelength UV/Vis band, correlate reasonably well with the electrochemical gaps Δ(Eox,1 − Ered,1), suggesting that the same orbitals are involved in the optical and electrochemical gaps for 1–7 (Table S1, ESI†). Furthermore, despite the different donor strengths, the electrochemical and optical HOMO–LUMO gaps are quite similar for all chromophores1–7, indicating that the first reduction takes place at the inner dicyanovinyl moieties and that the aniline moieties in these strong push–pull systems no longer act as strong donors, which is also supported by the similar high oxidation potentials of these moieties.
Inspired by the strongly anodically shifted reduction potentials, the electron-accepting power, expressed as adiabatic electron affinity (EA), was calculated at the (BP86/def-TZVP COSMO(ε = 4.5)//BP86/def-SV(P)) level19 for both conformers of acceptor 5. Despite the substitution with two strong diisopropylanilino donors, the predicted EAs for both the compact conformation (4.37 eV) and the extended geometry (4.63 eV) of 5 reach almost the value calculated for the state-of-the-art p-type dopant F4-TCNQ (4.96 eV),9a which suggests future exploration of the donor-substituted octacyano[4]dendralenes for opto-electronic device applications and solar cells.
Magnetic properties
The 1H NMR spectra of 5 in CD3CN (200 MHz) reveal strong line broadening at 20 °C. Upon raising the temperature to 70 °C, the lines become substantially narrower (see Fig. S1, ESI†). Significantly, all lines in the NMR spectrum are broadened to the same amount indicating that this is not due to dynamic effects, e.g. a hindered rotation about a single bond. It is more likely that this observation is based on the presence of paramagnetic, charge-separated stages of 5. Therefore, a sample used for the NMR measurements was placed into an EPR spectrometer. Here, a distinct but unresolved EPR spectrum could be recorded. Its intensity decreases with increasing the temperature (see Fig. S2, ESI†), perfectly reflecting the NMR behaviour and can be rationalised by a higher amount of the paramagnetic stage being present at lower temperatures. This points to a charged (or charge-separated) species, which is stabilised under more polar conditions, giving rise to the EPR signal.22a Although the EPR spectrum is unresolved, its width of ca. 2 mT and its g factor of 2.0030 are characteristic for a delocalised organic radical anion in which the spin population mainly resides at cyano-group containing moieties;22b moreover, such radical anions often lead to unresolved EPR spectra.9a Taking into account the LUMO of 5 (see Fig. S3, ESI†), which describes the electron distribution in 5 upon one-electron reduction, the recorded spectrum can tentatively be assigned to the radical anion of 5.Importantly, parent, solid 5 gives rise to an EPR signal (see Fig. S4, ESI†) compatible with 5•−. To estimate the amount of the content of the paramagnetic species, the magnetic susceptibility of solid 5 was measured. At room temperature, a very low value of χm = –2.16 × 10−9m3 kg−1 was determined, indicating that the sample is essentially diamagnetic. However, this value which depends on the magnetic field strength, reveals paramagnetic contributions underpins the observations made by NMR and EPR.
Conclusions
A new family of push–pull chromophores with a [4]dendralene backbone was synthesised by double [2+2] cycloaddition of TCNE to dianilino-end-capped buta-1,3-diynes, followed by retro-electrocyclisation. Despite the non-planarity of these systems, as revealed by X-ray crystallographic analyses, strong intramolecular charge-transfer (CT) interactions are effective. Two probable conformations, a compact and an extended one, were identified for these compounds in joined experimental and computational studies. They differ from each other primarily in the torsion angle for rotation about the central C–C bond of the [4]dendralene backbone. Gas-phase calculations show a preference for an extended geometry but the magnitude of this preference is dependent on environmental polarity. In the solid state, the compact conformation with all CN groups converging into one hemisphere is preferred: dimers are formed which are substantially stabilized by favourable intermolecular antiparallel dipolar CN⋯CN interactions. Electrochemical investigations by CV and RDV in CH2Cl2 reveal a remarkably high propensity for reversible electron uptake by these new chromophores, and calculations of the adiabatic electron affinity suggest a possible application of these molecules as p-type dopants in opto-electronic devices. The extension of this novel double cycloaddition/retro-electrocyclisation cascade to other acceptors, such as TCNQ and F4-TCNQ, and to differently activated oligoynes is being pursued.Acknowledgements
This work was supported by grants from the ETH Research Council, the European Research Council (ERC Advanced Grant No. 246637 (“OPTELOMAC”), the Austrian Science Fund (P20019) and by a doctoral stipend from the Stipendienfonds der Schweizerischen Chemischen Industrie (SSCI; Y.-L. W.) and a Kekulé Fellowship from the Fonds der Chemischen Industrie (B. B.). We are grateful for access to the Competence Center for Computational Chemistry (C4) Obelix cluster (ETH) and thank Prof. K. Gatterer (TU Graz) for carrying out the magnetic-susceptibility measurement.Notes and references
- For selected reviews on cyano-based acceptors, see: (a) T. L. Cairns and B. C. McKusick, Angew. Chem., 1961, 73, 520–525 CrossRef CAS; (b) O. W. Webster, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 210–221 CrossRef CAS.
- T. L. Cairns, R. A. Carboni, D. D. Coffman, V. A. Engelhardt, R. E. Heckert, E. L. Little, E. G. McGeer, B. C. McKusick and W. J. Middleton, J. Am. Chem. Soc., 1957, 79, 2340–2341 CrossRef.
- For selected reviews on the chemistry of TCNE, see: (a) A. J. Fatiadi, Synthesis, 1986, 249–284 CrossRef CAS; (b) A. J. Fatiadi, Synthesis, 1987, 749–789 CrossRef CAS; (c) A. J. Fatiadi, Synthesis, 1987, 959–978 CrossRef CAS; (d) J. S. Miller, Angew. Chem., Int. Ed., 2006, 45, 2508–2525 CrossRef CAS.
- (a) D. S. Acker, R. J. Harder, W. R. Hertler, W. Mahler, L. R. Melby, R. E. Benson and W. E. Mochel, J. Am. Chem. Soc., 1960, 82, 6408–6409 CrossRef CAS; (b) D. S. Acker and W. R. Hertler, J. Am. Chem. Soc., 1962, 84, 3370–3374 CrossRef CAS.
- For selected reviews on the chemistry of TCNQ, see: (a) B. P. Bespalov and V. V. Titov, Russ. Chem. Rev., 1975, 44, 1091–1108 Search PubMed; (b) W. Kaim and M. Moscherosch, Coord. Chem. Rev., 1994, 129, 157–193 CrossRef CAS.
- K. Walzer, B. Maennig, M. Pfeiffer and K. Leo, Chem. Rev., 2007, 107, 1233–1271 CrossRef CAS.
- (a) K.i. Onuma, Y. Kai, N. Yasuoka and N. Kasai, Bull. Chem. Soc. Jpn., 1975, 48, 1696–1700 CAS; (b) M. I. Bruce, J. R. Rogers, M. R. Snow and A. G. Swincer, J. Chem. Soc., Chem. Commun., 1981, 271–272 RSC.
- T. Michinobu, J. C. May, J. H. Lim, C. Boudon, J.-P. Gisselbrecht, P. Seiler, M. Gross, I. Biaggio and F. Diederich, Chem. Commun., 2005, 737–739 RSC.
- (a) M. Kivala, C. Boudon, J.-P. Gisselbrecht, B. Enko, P. Seiler, I. B. Mueller, N. Langer, P. D. Jarowski, G. Gescheidt and F. Diederich, Chem.–Eur. J., 2009, 15, 4111–4123 CrossRef CAS; (b) for a recent review, see: S.-i. Kato and F. Diederich, Chem. Commun., 2010, 46, 1994–2006 Search PubMed.
- (a) C. Koos, P. Vorreau, T. Vallaitis, P. Dumon, W. Boggerts, R. Baets, B. Esembeson, I. Biaggio, T. Michinobu, F. Diederich, W. Freude and J. Leuthold, Nat. Photonics, 2009, 3, 216–227 CrossRef CAS; (b) T. Vallaitis, S. Bogatscher, L. Alloatti, P. Dumon, R. Baets, M. L. Scimeca, I. Biaggio, F. Diederich, C. Koos, W. Freude and J. Leuthold, Opt. Express, 2009, 17, 17357–17368 CrossRef CAS . In both investigations, DDMEBT (see main text) was the NLO material deposited from the vapour phase.
- P. Reutenauer, M. Kivala, P. D. Jarowski, C. Boudon, J.-P. Gisselbrecht, M. Gross and F. Diederich, Chem. Commun., 2007, 4898–4900 RSC.
- For dendralenes, see: (a) M. Gholami and R. R. Tykwinski, Chem. Rev., 2006, 106, 4997–5027 CrossRef CAS; (b) H. Hopf, Angew. Chem., Int. Ed., 2001, 40, 705–707 CrossRef CAS; (c) A. D. Payne, G. Bojase, M. N. Paddon-Row and M. S. Sherburn, Angew. Chem., Int. Ed., 2009, 48, 4836–4839 CrossRef CAS.
- For a push–pull-substitued [4]dendralene, see: A. L. Kanobolotsky, J. C. Forgie, G. J. McEntee, M. M. A. Talpur, P. J. Skabara, T. D. J. Westgate, J. J. W. McDouall, M. Auinger, S. J. Coles and M. B. Hursthouse, Chem.–Eur. J., 2009, 15, 11581–11593 Search PubMed.
- M. I. Bruce, B. D. Kelly, B. W. Skelton and A. H. White, J. Organomet. Chem., 2000, 604, 150–156 CrossRef CAS.
- T. Michinobu, C. Boudon, J.-P. Gisselbrecht, P. Seiler, B. Frank, N. N. P. Moonen, M. Gross and F. Diederich, Chem.–Eur. J., 2006, 12, 1889–1905 CrossRef CAS.
- Mercury CSD 2.0: New Features for the Visualization and Investigation of Crystal Structures: C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 Search PubMed.
- For a review on multipolar interactions, see: R. Paulini, K. Müller and F. Diederich, Angew. Chem., Int. Ed., 2005, 44, 1788–1805 Search PubMed.
- P. A. Wood, S. J. Borwick, D. J. Watkin, W. D. S. Motherwell and F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2008, 64, 393–396 CrossRef.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- (a) Gaussian and Lorentzian curve fitting is not conclusive; (b) CT bands extending into the NIR region with weak extinction coefficients are observed in homoconjugated push–pull systems: S.-i. Kato, M. T. Roberts, P. Laporta, W. B. Schweizer, C. Boudon, J.-P. Gisselbrecht, I. Biaggio and F. Diederich, Angew. Chem., Int. Ed., 2010, 49, 6207–6211 Search PubMed.
- (a) S. Richeter, C. Jeandon, J.-P. Gisselbrecht, R. Ruppert and H. J. Callot, Inorg. Chem., 2007, 46, 10241–10251 CrossRef CAS; (b) K. M. Kadish, J. Shao, Z. Ou, C. P. Gros, F. Bolze, J. M. Barbe and R. Guilard, Inorg. Chem., 2003, 42, 4062–4070 CrossRef CAS; (c) R. Guilard, C. P. Gros, F. Bolze, F. Jérôme, Z. Ou, J. Shao, J. Fisher, R. Weiss and K. M. Kadish, Inorg. Chem., 2001, 40, 4845–4855 CrossRef CAS.
- (a) F. Gerson and W. Huber, Electron Spin Resonance Spectroscopy of Organic Radicals, Wiley-VCH, Weinheim, 2003, ch. 6.3, pp. 103–105 Search PubMed; (b) F. Gerson and W. Huber, Electron Spin Resonance Spectroscopy of Organic Radicals, Wiley-VCH, Weinheim, 2003, ch. 9.2, pp. 326–332 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Syntheses and UV/Vis spectra for new compounds, crystal structures of 5 and 7, variable-temperature 1H NMR and EPR spectra of 5 and computational details. CCDC reference numbers 775678 and 775679. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00387e |
| ‡ Crystal data for 5 (CCDC 775678) at 173 K: Single crystals were obtained by slow diffusion of hexane into a CH2Cl2 solution of 5 at 20 °C: C40H36N10·0.25CH2Cl2, M = 669.167, monoclinic, space groupP21/n (no. 14), a = 13.6680(6), b = 12.3175(6), c = 24.274(1) Å, β = 100.887(2)°, V = 4013.1(3) Å3, T = 173 K, Z = 4, 10888 reflections measured, 6241 unique (Rint = 0.112), R(F2) = 0.075 and wR(F2) = 0.174 on 3837 reflections with F2 > 2σ(F2).Crystal data for 7 (CCDC 775679) at 123 K: Single crystals were obtained by slow diffusion of hexane into a CH2Cl2 solution of 7 at 20 °C: C52H60N10, M = 825.122, monoclinic, space groupP21/c (no. 14), a = 14.1265(2), b = 21.8947(4), c = 15.3826(3) Å, β = 93.6655(9)°, V = 4748.0(1) Å3, T = 123 K, Z = 4, 49193 reflections measured, 10824 unique (Rint = 0.127), R(F2) = 0.064 and wR(F2) = 0.173 on 7710 reflections with F2 > 2σ(F2). |
| This journal is © The Royal Society of Chemistry 2011 |