Anodic deposition of a robust iridium-based water-oxidation catalyst from organometallic precursors†
James D.
Blakemore
a,
Nathan D.
Schley
a,
Gerard W.
Olack
b,
Christopher D.
Incarvito
a,
Gary W.
Brudvig
*a and
Robert H.
Crabtree
*a
aYale University, Department of Chemistry, PO Box 208107, New Haven, Connecticut 06520-8107, USA. E-mail: robert.crabtree@yale.edu; Tel: +1 203 432-3925; Fax: +1 203 432-6144; gary.brudvig@yale.edu; Tel: +1 203 432-5202; Fax: +1 203 432-6144
bYale University, Department of Geology and Geophysics, PO Box 208109, New Haven, Connecticut 06520-8109, USA
First published on 28th October 2010
Abstract
Artificial photosynthesis, modeled on natural light-driven oxidation of water in Photosystem II, holds promise as a sustainable source of reducing equivalents for producing fuels. Few robust water-oxidation catalysts capable of mediating this difficult four-electron, four-proton reaction have yet been described. We report a new method for generating an amorphous electrodeposited material, principally consisting of iridium and oxygen, which is a robust and long-lived catalyst for water oxidation, when driven electrochemically. The catalyst material is generated by a simple anodic deposition from Cp*Ir aqua or hydroxo complexes in aqueous solution. This work suggests that organometallic precursors may be useful in electrodeposition of inorganic heterogeneous catalysts.
Introduction
Water-oxidation catalysts are envisioned to play a key role in many proposed schemes for artificial photosynthesis and solar-fuel production.1 In these proposals, photoelectrochemical cells harvest and store solar energy in chemical form. Although details vary, the strategy typically proposed is the oxidation of water at an anode to produce electrons that in turn reduce H2O or CO2 to fuels at the cell cathode.The high kinetic barrier for oxygen evolution is a key hurdle in each of these proposed systems, since it limits cell power efficiency by introducing a large overpotential.2 Additionally, the required oxidizing equivalents must be properly coupled to yield dioxygen, rather than partially oxidized side products (e.g., HO˙, H2O2).3 These difficulties hamper the development of water-oxidation catalysts, especially catalysts that can operate under the required harsh oxidizing conditions in a sustained fashion with minimal deactivation.
The dependence of overpotential for oxygen evolution on the nature of the electrode material has been recognized for many years.4 Noble metal oxides (e.g., ruthenium, iridium) were observed early to have good catalytic properties for water oxidation.5 Among these, iridium materials have proven to be among the most active and stable for oxygen evolution.6 The advent of Dimensionally Stable Anodes® (DSA) in the 1960s introduced iridium oxides as anodes in industrial applications requiring high activity and longevity.7 These electrodes are composed of an oxidation resistant titanium/titanium oxide substrate which is coated with an active catalyst layer. This active layer often includes several metal oxides (e.g., tantalum, titanium) in addition to iridium, in order to disperse the active catalyst material, provide high surface area, and reduce corrosion.8 However, the catalyst for the oxygen evolution reaction is the iridium material.
Iridium oxides for use as electrode materials have historically been prepared in three ways. In the first, a solution containing iridium chlorides is applied to the electrode surface and then thermally decomposed, giving a nanoparticulate material which is an active catalyst.9 In the second method, thermal or chemical decomposition of iridium salts under controlled solution conditions gives rise to free nanoparticles, which can be physically or electrochemically deposited on the electrode surface.10,11 Chemical preparations of this second type are used in proton-exchange membrane (PEM) water electrolyzers.12 Sputtered iridium dioxide has also been shown to be highly active for oxygen evolution.13 Additionally, various types of iridium oxide films have been previously shown to be electrochromic, with the most active prepared by the sputtering technique.14
In more recent work, we have shown that a number of organometallic pentamethylcyclopentadienyl (Cp*) iridium complexes are robust and highly active homogeneous catalysts for water oxidation when driven with cerium(IV) as primary oxidant.15 Complexes 1 and 2 (Fig. 1),16 having multiple aqua or hydroxo ligands show the fastest rates of oxygen evolution, up to 20–25 turnovers min−1 even at pH 0.89 with cerium(IV).‡,17 These values compare well with recently published work on cobalt catalysis.18 Kinetic studies, reported elsewhere,17 show that the order of reaction in iridium for complexes 1 and 2 is greater than one, suggesting dimerization or oligomerization is key to forming the catalyst of highest activity.
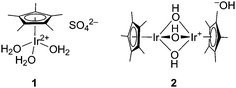 | ||
| Fig. 1 Structures of iridium(III) complexes 1 and 2. | ||
We now report that electrochemical oxidation of 1 or 2 induces deposition of a catalytic blue layer of an iridium oxide material (BL) capable of catalyzing water oxidation at the anode. Holding the oxidizing potential steady for several minutes is sufficient to form a visible amount of BL. Optimal activity is reached after deposition for several hours (see ESI†). BL deposits on carbon, platinum, gold, fluorine-doped tin oxide (FTO), and indium tin oxide electrodes. We know of no prior case in which an active material for water oxidation has been deposited from a well-defined organometallic precursor having a metal–carbon bond, although inorganic salts and molecular coordination complexes have been used previously for several metals including cobalt and nickel.19 Importantly, this route is direct and simple, avoiding harsh conditions and specialized equipment. Furthermore, the deposited BL displays interesting electrochromic properties, similar to classical sputtered iridium oxides.
Results and discussion
A cyclic voltammogram of a solution containing 1 (Fig. 2, inset) shows only a simple catalytic wave on the first anodic scan with onset at 1.15 V (all potentials are vs. NHE). On multiple cycling, the BL builds up, accompanied by a feature at 0.88 V that grows in intensity. As more BL deposits, the catalytic wave at 1.1 V also becomes more intense.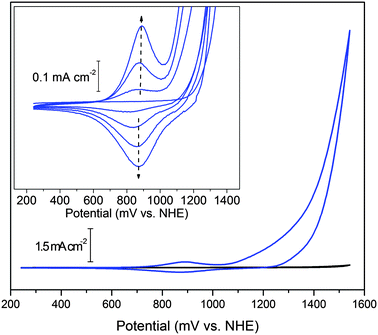 | ||
| Fig. 2 Cyclic voltammetry of BL (blue line) and basal plane graphite electrode background (black line) in 0.1 M KNO3 at pH 6. The catalytic wave is seen above ca. 1.1 V. Inset: cyclic voltammogram (successive scans) showing deposition of BL from a solution containing 1; reversible peak centered at 0.88 V (ΔEp = 15 mV). Deposition conditions: 2.3 mM 1, 0.1 M KNO3, pH 2.9; scan rate: 50 mV s−1. | ||
Upon rinsing the anode and transferring it to a solution of pure potassium nitrate electrolyte, the reversible feature and catalytic wave from the BL are still present at full intensity. The peak separation, ΔEp, for the reversible feature is ca. 15 mV, consistent with a non-diffusional, surface-bound species (ideally, ΔEp = 0). Peak currents for the reversible feature show a linear dependence with scan rate, as expected for a surface-bound species (see ESI†).20 Thus, the BL is deposited at the electrode surface and is responsible for the catalytic reaction.§
Upon deposition, BL functions as a robust and highly active catalyst for water oxidation. Indeed, bubbles of O2 are rapidly formed at the electrode surface during cyclic voltammetry. Oxygen was confirmed as the product of the catalytic oxidation wave by headspace gas analysis using a fluorescence-probe assay (Fig. 3). An FTO slide, previously prepared with BL, was introduced into a closed two-chamber electrochemical cell with 0.1 M KNO3 (pH 7), and 1.4 V was applied for 1 h. During this time, sufficient charge was passed through the cell to generate 28.9 μmol of dioxygen. 27.7 μmol was detected by the headspace oxygen assay, representing a Faradaic efficiency of 96%. In Fig. 3, it is clear from the parallel behavior of the two signals that nearly all of the oxidizing equivalents contribute to oxygen evolution, rather than to detrimental side reactions. Water was confirmed as the source of oxygen atoms by H218O isotope incorporation to give the expected 18O enrichment in product O2 (see ESI†).
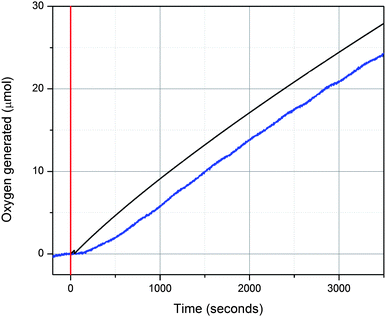 | ||
| Fig. 3 Oxygen evolution catalyzed by BL. Black line: predicted oxygen yield based on current transferred. Blue line: oxygen yield detected by headspace analysis. | ||
BL is resistant to corrosion and to loss of catalyst during operation. Long time course studies indicate that activity is retained over many hours of continuous use. Specifically, after 9 h of constant electrolysis at an applied potential of 1.4 V (sustained current density: 1.4 mA cm−2, without cell resistance (iR) compensation), the catalyst maintains over 95% of its initial activity (see ESI†).
BL also exhibits electrochromism. If the electrode is polarized at a potential below the reversible wave centered at 0.88 V, the film becomes largely translucent, turning a very pale shade of yellow–green. Not surprisingly, polarizing the electrode above the reversible wave causes the striking blue color and opacity to return. This color change also occurs on treatment of BL with chemical reductants and oxidants, consistent with an oxidation state change from predominantly iridium(III) to predominantly iridium(IV), similar to results on iridium films prepared by other methods.21
The reversible wave at 0.88 V can be used to estimate the number of surface-bound electroactive iridium atoms. In previously studied iridium oxide films, the reversible wave was anticipated to be a one-electron process (iridium(III) to iridium(IV)). However, careful studies by Burke and Whelan led to the observation that the reversible wave is actually a two-electron, three-proton process.22 Based on the similarity of BL in voltammetric and catalytic response to the previously described hydrous iridium oxide films, the assumption of a two-electron, three-proton process is used here. After 10 or 20 cycles from 0.3 to 1.5 V at 100 mV s−1, the surface coverage of iridium was estimated to be 4.1 and 8.2 nmol cm−2, respectively. The former can be estimated at a coverage of less than ten monolayers, in analogy with similar work previously carried out with Au(111) monolayers.20
The turnover frequency on a per-electroactive iridium basis can be estimated with the surface coverage data. At the coverages listed above, the turnover frequency at 1.4 V was estimated to be 1.46 turnovers s−1 for the 4.1 nmol cm−2BL and 1.11 turnovers s−1 for the 8.2 nmol cm−2BL (platinum disc rotating disc electrode, ω = 1000 rpm). This is lower than the turnover values reported by Murray, et al., for small iridium dioxide nanoparticles at 1.5 V.11b Furthermore, some surface-bound iridium atoms may not be electroactive, complicating this analysis. In a highly active film which has been deposited for 2 h, approximately 0.25 mg cm−2 is present on the electrode surface; however, at these loadings, the features in cyclic voltammetry are broadened so significantly that no meaningful analysis can be accomplished. Films of this type have a thickness of between 1 and 2 μm, as estimated by SEM. (see ESI†)
In general, the discrepancy between measured activity and the apparent robustness of catalysis may be explained by a non-Nernstian response from the BL. The full width at half maximum measured for the reversible feature at 0.88 V is consistently much larger than predicted by electrochemical theory, even at low catalyst loadings (vide supra).15 Since this is the case, the turnover frequency on a total iridium basis cannot be confidently estimated.
The high activity of the BL presumably results from its high surface area. Indeed SEM images show nanoscale features, consistent with this hypothesis (Fig. 4). However, the deposited material is amorphous by X-ray diffraction (only broad, low-angle diffraction is seen; see ESI†).

Elemental analysis on BL (after depositing at 1.4 V for 2 h) shows 9% carbon by mass, along with hydrogen, oxygen and iridium. There is negligible nitrogen content.¶ The results are consistent with energy-dispersive X-ray spectroscopy (EDS) measurements, which show the presence of only carbon, oxygen and iridium above background after deposition (see ESI†). BL samples deposited from solutions of 1 do not show sulfur incorporation by EDS. As Cp* is the only source of carbon, we conclude that a substoichiometric fraction of the Cp* ligand, or a derivative, is incorporated into BL. The Cp* ligand is necessary for deposition, as solutions of iridium trichloride hydrate in potassium nitrate do not form deposits or show catalytic oxidation waves in cyclic voltammetry, nor do less coordinatively unsaturated Cp*Ir complexes such as Cp*Ir(κ2-2-phenylpyridine)Cl or Cp*Ir(2,2′-dipyridyl)Cl2. Oxidation of Cp* methyl groups has been observed previously on treatment with iodine(III) reagents.23 Similar oxidation could contribute to the deposition process for BL, but complexes 1 and 2 are the ones that form a deposit, in contrast to the other half-sandwich iridium compounds we have studied.
IR spectra collected for BL also show clear indications of carbon-containing fragments (see ESI†). There are broad, strong stretching frequencies in the region of 1300–1700 cm−1, assigned to ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and ν(C–O) features, which would arise upon oxidative degradation the Cp* ligand. However, there are no detectable ν(C–H) frequencies, indicating that essentially all of the Cp* methyl groups have been lost upon formation of BL.
O) and ν(C–O) features, which would arise upon oxidative degradation the Cp* ligand. However, there are no detectable ν(C–H) frequencies, indicating that essentially all of the Cp* methyl groups have been lost upon formation of BL.
We next examined the overpotential for oxygen evolution. Bubble formation and large local pH effects, possible during electrolysis, seem to complicate measurements; therefore, employing a rotating-disc electrode allows us to conduct the experiment under controlled conditions. However, because the Tafel plot (Fig. 5) is not linear, the overpotential cannot be assessed here with complete reliability. Nonetheless, the data show the overpotential required to reach 0.5 mA cm−2 is 200 ± 3 mV. In fact, due to the highly capacitative nature of hydrated noble metal oxides, this overpotential will likely be underestimated (see ESI†). Comparison with very long timecourse data collected without a rotating disc electrode (9 h data from above) at 1.4 mA cm−2 shows a near 100 mV higher overpotential, although the rotating disc experiments do not suffer from bubble formation and consequent loss of surface area. Generally, these experiments show there is a low overpotential (≤300 mV) for water oxidation with BL, demonstrating the dramatic lowering of the kinetic barrier to OO bond formation achieved by catalysis with BL.
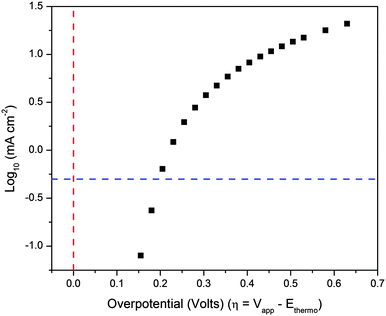 | ||
| Fig. 5 Tafel plot of catalytic currents. Blue dashed line corresponds to 0.5 mA cm−2; red dashed line is at zero overpotential. Conditions: 0.1 M KNO3, pH 2.9, platinum rotating disc electrode. | ||
Discussion
Iridium oxide materials are known to be highly active catalysts for water oxidation.5,6 With this in mind, it is not surprising that formation of BL on the electrode surface coincides with the appearance of a catalytic response leading to water oxidation. There are striking similarities in electrocatalytic behavior (i.e., low overpotential) and voltammetric response (i.e., appearance of cyclic voltammograms, electrochromism) between BL and other preparations of iridium oxides.24 This strongly suggests that inorganic iridium dioxide is the active catalyst in this case, especially considering the strong effect of preparation method on heterogeneous catalyst performance. However, unlike many other preparations of iridium oxide, BL is prepared by an exceptionally mild method. Preparation of the catalyst from organometallic precursors provides a route to explore new solid-state and inorganic catalytic materials for water oxidation. Further, as iridium oxides are one of the most active and industrially-relevant materials available today for water oxidation chemistry, this novel method of preparation will help in elucidating their mechanism of action and perhaps lead to significant improvements in catalyst performance. Specifically, since BL is prepared by a mild and facile method, it may be of use in assemblies for achieving light-driven water oxidation.25Conclusions
In conclusion, we report the iridium oxideBL is a highly active and robust electrocatalyst for water oxidation. It is prepared by electrodeposition from aqueous solutions of organometallic Cp*Ir precursors, allowing careful control of catalyst loading and preparation conditions. The activity of BL suggests that it is a promising platform for investigating catalyticwater oxidation at electrode surfaces. Furthermore, the method of preparing BL from an organometallic precursor suggests that a new, diverse range of heterogeneous catalysts may be prepared from solutions of organometallic precursors, perhaps eventually with abundant first-row metals.Acknowledgements
This material is based upon work supported as part of the Argonne-Northwestern Solar Energy Research (ANSER) Center, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-PS02-08ER15944 (G. W. B., R. H. C., and J. D. B.). Further funding from the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences of the U.S. Department of Energy through grant DE-FG02-84ER13297 (R. H. C. and N. D. S.) is gratefully acknowledged. The authors thank the referees for their helpful comments. J. D. B. thanks Graham Dobereiner and Jonathan Parr for helpful discussions. Isotope incorporation experiments were carried out at the Earth Systems Center for Stable Isotope Studies at Yale University.Notes and references
- (a) T. J. Meyer, Acc. Chem. Res., 1989, 22, 163–170 CrossRef CAS; (b) A. J. Bard and M. A. Fox, Acc. Chem. Res., 1995, 28, 141–145 CrossRef CAS; (c) X. Sala, I. Romero, M. Rodríguez, L. Escriche and A. Llobet, Angew. Chem., Int. Ed., 2009, 48, 2842–2852 CrossRef CAS.
- D. G. H. Hetterscheid, J. I. van der Vlugt, B. de Bruin and J. N. H. Reek, Angew. Chem., Int. Ed., 2009, 48, 8178–8181 CrossRef CAS.
- J. O'M. Bockris, Energy: The Solar-Hydrogen Alternative, Hogbin and Poole, Australia, 1975 Search PubMed.
- A. Hickling and S. Hill, Discuss. Faraday Soc., 1947, 1, 236–246 Search PubMed.
- (a) A. Harriman, M.-C. Richoux, P. A. Christensen, S. Mosseri and P. Neta, J. Chem. Soc., Faraday Trans. 1, 1987, 83, 3001–3014 RSC; (b) A. Harriman and J. M. Thomas, New J. Chem., 1987, 11, 757–762 CAS.
- X. Chen, G. Chen and P. L. Yue, J. Phys. Chem. B, 2001, 105, 4623–4628 CrossRef CAS.
- (a) H. B. Beer, J. Electrochem. Soc., 1980, 127, 303C–307C CAS; (b) P. C. S. Hayfield, Platinum Met. Rev., 1998, 42, 116–122 CAS.
- (a) Ch. Comninellis and G. P. Vercesi, J. Appl. Electrochem., 1991, 21, 335–345 CrossRef CAS; (b) S. Trasatti, Electrochim. Acta, 2000, 45, 2377–2385 CrossRef CAS.
- Ch. Comninellis and G. P. Vercesi, J. Appl. Electrochem., 1991, 21, 136–142 CAS.
- K. Yamanaka, Jpn. J. Appl. Phys., 1989, 28, 632–637 CrossRef CAS.
- (a) M. A. Petit and V. Plinchon, J. Electroanal. Chem., 1998, 444, 247–252 CrossRef CAS; (b) T. Nakagawa, C. A. Beasley and R. W. Murray, J. Phys. Chem. C, 2009, 113, 12958–12961 CrossRef CAS.
- P. Millet, R. Durand and M. Pineri, Int. J. Hydrogen Energy, 1990, 15, 245–253 CrossRef CAS.
- G. Beni, L. M. Schiavone, J. L. Shay, W. C. Dautremont-Smith and B. S. Schneider, Nature, 1979, 282, 281–283 CrossRef CAS.
- K. S. Kang and J. L. Shay, J. Electrochem. Soc., 1983, 130, 766–769 CrossRef CAS.
- J. F. Hull, D. Balcells, J. D. Blakemore, C. D. Incarvito, O. Eisenstein, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2009, 131, 8730–8731 CrossRef CAS.
- (a) S. Ogo, N. Makihara and Y. Watanabe, Organometallics, 1999, 18, 5470–5474 CrossRef CAS; (b) A. Nutton, P. M. Bailey and P. M. Maitlis, J. Chem. Soc., Dalton Trans., 1981, 1997–2002 RSC.
- J. D. Blakemore, N. D. Schley, D. Balcells, J. F. Hull, G. W. Olack, C. D. Incarvito, O. Eisenstein, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2010 Search PubMed , in press.
- Q. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342–345 CrossRef CAS.
- (a) M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS; (b) M. Dinca, Y. Surendranath and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10337–10341 CAS.
- A. J. Bard, and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, 2nd edn, Wiley, Hoboken, 2001 Search PubMed.
- L. D. Burke and R. A. Scannell, Platinum Met. Rev., 1984, 28, 56–61 CAS.
- (a) L. D. Burke and D. P. Whelan, J. Electroanal. Chem., 1981, 124, 333–337 CrossRef CAS; (b) L. D. Burke and D. P. Whelan, J. Electroanal. Chem., 1984, 162, 121–141 CrossRef CAS.
- L. S. Park-Gehrke, J. Freudenthal, W. Kaminsky, A. G. DiPasquale and J. M. Mayer, Dalton Trans., 2009, 1972–1983 RSC.
- S. Gottesfeld and S. Srinivasan, J. Electroanal. Chem., 1978, 86, 89–104 CrossRef CAS.
- P. D. Tran, V. Artero and M. Fontecave, Energy Environ. Sci., 2010, 3, 727–747 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Powder X-ray diffraction data. SEM and EDS data. Additional electrochemical characterization. Oxygen evolution and 18O isotope incorporation details. Detailed experimental procedures. See DOI: 10.1039/c0sc00418a |
| ‡ Turnovers are defined as: mol O2 (mol Ir)−1. |
| § Under the deposition conditions, the local pH at the electrode surface was anticipated to be lower than in the bulk solution, due to proton release during vigorous water oxidation. This would cause 2 to protonate, essentially forming 1in situ. Investigations are now underway to elucidate differences between BL deposited from 1vs. 2. |
| ¶ Elemental analysis results for C, N, H, O and Ir sum to only 74%. This is likely due to a low recovery during analysis, a common occurrence with metal oxide materials. Analysis performed by Columbia Analytical Services, Inc., 3860 S. Palo Verde Rd. Ste. 302, Tucson, Arizona 85714. |
| This journal is © The Royal Society of Chemistry 2011 |