Catalytic asymmetric α-alkylation of aldehydesvia a SN2′-type addition-elimination pathway†
Enrique
Gómez-Bengoa
,
Aitor
Landa
,
Aitziber
Lizarraga
,
Antonia
Mielgo
,
Mikel
Oiarbide
and
Claudio
Palomo
*
Departamento de Química Orgánica I, Universidad del País Vasco, Apdo. 1072, 20080, San Sebastián, Spain. E-mail: claudio.palomo@ehu.es; Fax: (+) 943015270
First published on 25th October 2010
Abstract
A novel method for the direct, amine-catalyzed, highly enantioselective α-alkylation of aldehydes is described that is founded upon the use of electrondeficient allylic halides as alkylating agents and DABCO or DMAP as coadyuvant. Both experimental observations and DFT calculations, in support of a SN2′-type addition-elimination pathway involving ammonium salt intermediates, are provided.
Introduction
Alkylation of carbonyl compounds at the α carbon upon reaction with an alkyl halide (or equivalent alkylating reagent) is one of the most popular C–C bond forming reactions in organic chemistry. Highly reliable asymmetric variants of this transformation have been established and applied in synthesis, particularly for substrates in the carboxylic acid oxidation state. Most often these methods involve preformation of a discrete metal enolate, which is then treated with the pertinent alkyl halide.1 However, metal enolates from aldehydes show promiscuous reactivity, leading to several competing undesired side reactions (self-aldol condensation, Cannizaro and Tischenko reactions, among others).2 Thus, the catalytic asymmetric α-alkylation of aldehydes remains a long-standing problem.3Recent progress in this area has been made using the potential of aminocatalysis and its adaptability to specific reaction mechanisms, from intramolecular SN2 to intermolecular radical, and SN1 pathways (Scheme 1).4 In 2004, Vignola and List reported α-methyl proline-catalyzed intramolecular reactions of ω-haloaldehydes to provide three and five member carbo- and heterocycles enantioselectively.5,6 More recent studies carried out independently by the groups of MacMillan,7 Melchiorre and Petrini,8 Cozzi,9 and Jacobsen,10 have not only provided novel methods for the aminocatalytic, asymmetric α-alkylation of aldehydes,11 but have also demonstrated the compatibility of enamine activation with radical and cationic (SN1) reaction pathways. These innovative methods represent a clear advance, but often require more sophisticated alkylating reagents/catalytic systems or are restricted to a certain class of alkylating electrophiles. In this Edge article, we describe an amine-catalyzed asymmetric α-alkylation of aldehydes that follows a distinct mechanism, based on the use of electrondeficient allylic bromides as alkylating reagents.
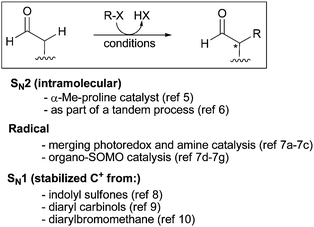 | ||
| Scheme 1 Current, mechanistically complementary, solutions to the catalytic asymmetric α-alkylation of unmodified aldehydes. | ||
Results and discussions
A major problem for the development of a general, amine-catalyzed, α-alkylation of aldehydes with alkyl halides is catalyst inactivation through (i) acid–base neutralization effected by the evolving acid HX, or (ii) competitive irreversible N–alkylation of the catalyst.5a,11b Problem (i) can be circumvented by carrying out the reaction in the presence of an acid scavenger (typically, a tertiary amine base), but problem (ii) does not have an obvious solution. Our assumption was that by using shoft alkylating reagents, such as the electrondeficient allyl halides of type A (Scheme 2), the undesired N–alkylation of the catalyst could be rendered reversible or marginal. Accordingly, a threshold concentration of free amine catalyst should remain in solution so as to keep operative the enamine-mediated irreversible C–C bond forming reaction to eventually provide the α-alkylated product B in a stereo-biased manner.12 In addition, the use of reagent A is particularly attractive in that functionalized products are attainable, as illustrated in recent enantioselective cinchona alkaloid-catalyzed alkylation reactions of active methylenes and indoles.13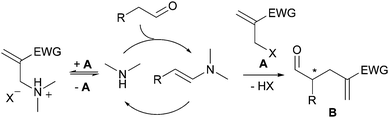 | ||
| Scheme 2 Amine-catalyzed α-alkylation of aldehydes with allyl halides A, assuming a reversible N–alkylation side reaction (EWG: electron withdrawing group). | ||
Initial attempts for experimental validation of the above hypothesis using 2-(halomethyl)acrylates unravelled two additional difficulties for this challenging transformation to overcome; namely (a) the inefficacy of simple acrylate esters towards the addition reaction of transient enamine species14 and (b) the known tendency of α-branched aldehydes to undergo base-promoted racemization. Thus, Scheme 3, attempted the reaction of hydrocinnamaldehyde 1a (R = CH2Ph) with allyl bromide 2, catalyzed by the chiral pyrrolidine 5a15 (20 mol %) in the absence of any base, or the presence of 1 equivalent triethylamine (TEA) or diisopropylethylamine (DIPEA) as acid scavengers, led to an unaltered starting aldehyde and halide (Table 1, entries 1–3).
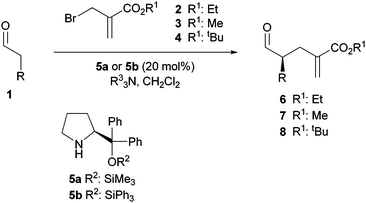 | ||
| Scheme 3 Direct enantioselective α-alkylation of aldehydes with 2-(bromomethyl) acrylates. | ||
| Entry | R33N | Cat | T/°C | t/h | Conv. (%)b | ee (%)c |
|---|---|---|---|---|---|---|
| a General conditions: 1a (1.5 mmol), 2 (0.5 mmol), R33N (0.51 mmol), catalyst 5 (20 mol %) were stirred in CH2Cl2 (2 mL) at the indicated temperature. b Determined by 1H NMR. c Determined by chiral HPLC of crude product before chromatography. NR: no reaction. | ||||||
| 1 | -- | 5a | 20 | 24 | NR | -- |
| 2 | TEA | 5a | 20 | 24 | NR | -- |
| 3 | DIPEA | 5a | 20 | 24 | NR | -- |
| 4 | DBU | 5a | 20 | 24 | >95 | 0 |
| 5 | DABCO | 5a | 20 | 24 | >95 | 81 |
| 6 | DMAP | 5a | 20 | 24 | >95 | 79 |
| 7 | DMAP | 5a | 0 | 24 | 95 | 85 |
| 8 | DMAP | 5b | 0 | 36 | 70 | 89 |
| 9 | DMAP | 5b | –10 | 48 | >95 | 93 |
Further screening of other stoichiometric amine bases eventually identified DBU as a successful promoter of the reaction, to afford the alkylated product 6a, albeit as a racemic product (entry 4). The null enantioselection observed was primarily attributed to the strong basicity of DBU in connection to problem (b). Most importantly, the dramatic effect in reactivity observed, suggested that the actual alkylating agent under these reaction conditions might likely be the corresponding ammonium salt derived from halide A and DBU,16 rather than the halide itself. Based on this assumption, the alkylation reaction was examined using DABCO and DMAP, which have been shown by Mayr to possess optimal nucleophilicity/nucleofugacity while being less basic than DBU.17 Gratifyingly, when the above reaction was carried out in the presence of 1 equivalent of either DABCO or DMAP, the alkylated aldehyde 6a was formed after 24 h at room temperature, with ee's of 81% and 79%, respectively (entries 5 and 6). Further improvement of the ee was attained by decreasing the reaction temperature (0 °C, entry 7) or by shifting to the catalyst 5b (entries 8 and 9).15c
Exploration of the scope of the reaction with respect to the aldehyde substrate, revealed satisfactory results for a range of aldehydes with varying steric demand and functionality, Table 2. Thus, aldehydes with linear alkyl chains of variable length afforded good yields and ee's uniformly above 90% (entries 1–6); slightly lower reactivity was shown by branched chain aldehydes, but still high selectivity was achieved (entry 7). Similarly, a range of functional groups, including alkene, ether, ester, acetal, and carbamate, are tolerated (entries 8–15), although the alkoxyaldehyde 1i gave diminished ee for reasons not yet well understood (entry 12). In general, DABCO affords similar or even slightly higher ee’s than DMAP at r.t. or 0 °C, but at lower temperatures DMAP showed superior ee’s for most cases.18 Interestingly, α-alkylation of the chiral aldehyde citronellal took place with remarkable diastereoselectivity for the matched pair (entry 11). On the other hand, control experiments using the reaction adducts were informative with respect to their configurational robustness against acidic or basic conditions. For example, the measured values of the ee before and after silica gel chromatography for adduct 6a were essentially identical. No appreciable variation of ee was either observed even after stirring a solution of 6a in CH2Cl2 admixed with silica gel at room temperature for 16 h. In turn, while exposure of 6a and 6d to DMAP or DABCO (5 mol%) at 0 °C overnight did not affect their configurational integrity, essentially complete racemization took place in the presence of DBU (5 mol%) after 2–3 h at the same temperature. A brief exploration of the influence of the size of the ester group on the reaction outcome, indicated that methyl ester 3 is equally suitable than the parent ethyl ester (compare entries 1 and 16); however, the tert-butyl ester 4 showed reduced reactivity at −10 °C to produce 8a, and at room temperature the reaction was feasible using either DMAP or DABCO, although at the expense of a selectivity decrease (entry 17).
| Entry | R | Product | T/°C | t/h | Yield (%)a | ee (%)b |
|---|---|---|---|---|---|---|
| a Yield of isolated product after chromatography. In parenthesis, results using DABCO instead of DMAP. b Determined by chiral HPLC. c Ratio of diastereomers. d Conversion after 52 h of reaction. | ||||||
| 1 | CH2Ph | 6a | 0 | 48 | 56 (45) | 90 (94) |
| 2 | –10 | 48 | 59 | 93 | ||
| 3 | CH2CH3 | 6b | 0 | 48 | 61 (50) | 86 (91) |
| 4 | –10 | 48 | 63 | 92 | ||
| 5 | (CH2)3CH3 | 6c | 0 | 48 | 64 (49) | 85 (92) |
| 6 | –10 | 48 | 61 | 94 | ||
| 7 | CH(CH3) 2 | 6d | 0 | 60 | 54 | 90 |
| 8 | CH2CH = CH2 | 6e | –20 | 60 | 63 | 95 |
| 9 | (CH2)2CH = CH2 | 6f | –20 | 60 | 69 | 93 |
| 10 | (R)-Citronellal | 6g | 20 | 48 | 68 | 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25c :25c |
| 11 | (S)-Citronellal | 6h | 20 | 48 | 70 | 97:3c |
| 12 | CH2OBn | 6i | –10 | 60 | 59 | 68 |
| 13 | (CH2)2CO2Me | 6j | –10 | 60 | 62 | 90 |
| 14 | CH2 CH(OMe)2 | 6k | –10 | 60 | 58 | 87 |
| 15 | (CH2)4NHBoc | 6l | –10 | 60 | 62 | 94 |
| 16 | CH2Ph | 7a | –10 | 60 | 57 | 95 |
| 17 | CH2Ph | 8a | 20 | 52 | 80d (90)d | 85 (79) |
The orthogonal functionality of the resulting alkylated adducts provides versatility to this catalytic methodology. For instance, Scheme 4, reduction of aldehyde 6e and smooth lactonization of the resulting alcohol, afforded a known compound 10,19 which served to establish the stereochemistry of the catalytic reactions. Further potential of the method could be inferred from the ring closing metathesis of adducts 6e and 6f, using Grubbs' catalyst 9,20 to yield the corresponding 5- and 6-membered carbocycles 11e and 11f, in high enantioselectivity.21
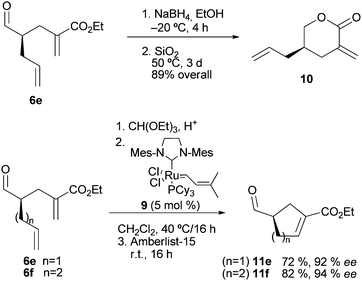 | ||
| Scheme 4 Elaboration of adducts into hetero- and carbocycles. | ||
Complementing these experiments, which serve as a proof of concept of the validity of halides of type A as suitable electrophiles for the difficult to control amine-catalyzed asymmetric α-alkylation of aldehydes,2,4 the reaction mechanism was briefly investigated computationally. In this respect, DFT calculations for the model reaction involving the methyl acrylate 3, DMAP, and the enamine derived from acetaldehyde and pyrrolidine, suggest an asynchronous SN2′ pathway for the initial step leading to the ammonium salt 12 (Fig. 1a), with the C–N bond formation preceding the C–Br bond breaking. For the subsequent reaction of 12 with the enamine, the energy profile (Fig. 1b) indicates a two step addition-elimination sequence, with 13 being the intermediate formed after the rate limiting addition step.23,24 The alternative direct displacement (SN2) pathway for both the C–N and C–C bond forming processes was also calculated, resulting in much higher energy barriers in both cases.25 Consistent with the above reaction scheme is the fact that treatment of butyraldehyde with the preformed ammonium salt from 2 and DMAP provided, in the presence of 20 mol% of catalyst 5b, alkylated adduct 6b in essentially the same yield and ee as in entry 3 of Table 2.
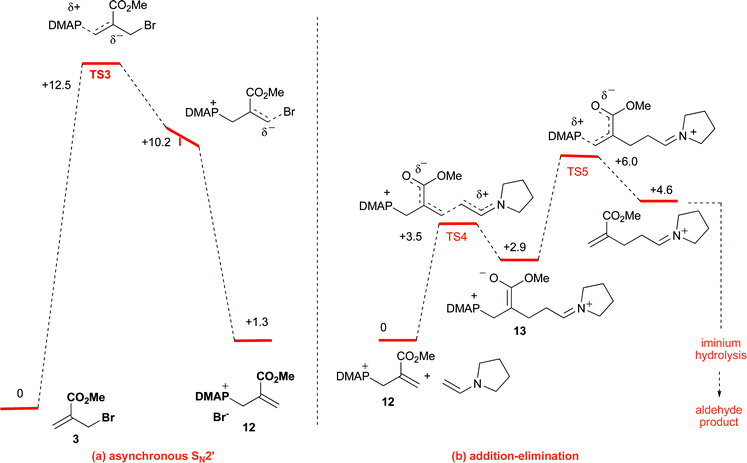 | ||
| Fig. 1 Reaction Coordinate Diagram for the model reaction computed at the B3LYP/6–31* level (enthalpies given in kcal mol−1) corresponding to: (a) initial formation of intermediate ammonium salt 12 (asynchronous SN2′); (b) key C–C bond forming step (two-step addition-elimination SN2′-type mechanism). | ||
Finally, it was found that substitution at the β-position of the 2-(bromomethyl)acrylate exerts a notable effect upon reactivity. As the example in Scheme 5 shows, the reaction of butyraldehyde with 14 in the presence of DMAP was extremely slow, but with DABCO proceeded to give product 15 in moderate yield after prolonged reaction time. Significantly, 15 was the only regioisomer detected, which was obtained in remarkable diastereo- and enantioselectivity.22
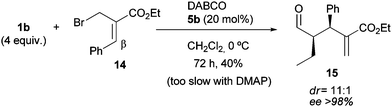 | ||
| Scheme 5 Regio-, diastereo-, and enantioselective α-alkylation of aldehydes with β-substituted α-bromomethyl acrylate 14. | ||
Conclusions
The present Edge article discloses the first enamine-mediated direct asymmetric α-alkylation of aldehydes, in which the key C–C bond forming event proceeds through a SN2′-type addition-elimination mechanism, thus complementing the known SN2, SN1, and radical approaches. This method (i) is operationally simple (ii) needs no metal containing reagent or catalyst, (iii) shows wide functional group compatibility, and (iv) provides a rapid entry to useful carbo- and heterocycles. The validity of the method with related allylic electrophiles, as well as further synthetic application of the alkylated adducts are under investigation and the results will be reported in due course.Acknowledgements
Financial support was provided by the University of the Basque Country (UPV/EHU), and Ministerio de Educación y Ciencia (MEC, Grant CTQ2007-68095-C02), Spain. A. L. thanks MEC and European Social Foundation for a Ramón y Cajal contract. A. L. thanks UPV/EHU for a fellowship. We also thank SGIker (UPV/EHU) for providing NMR, HRMS, and computational resources.Notes and references
- Houben-Weyl Methods of Organic Chemistry, Stereoselective Synthesis, ed. G. Helmchen, R. W. Hoffmann, J. Mulzer, E. Schaumann, Thieme, Stuttgart-New York, 1996, vol 2 (E 21) pp 697–1015 Search PubMed.
- (a) G. Stork, A. Brizzolara, H. Landesman, J. Szmuszkovicz and R. Terrell, J. Am. Chem. Soc., 1963, 85, 207–222 CrossRef CAS; (b) H. O. House, W. C. Liang and P. D. Weeks, J. Org. Chem., 1974, 39, 3102–3107 CrossRef CAS.
- (a) Modern Carbonyl Chemistry, ed. J. Otera, Wiley-VCH, Weinheim, 2000 Search PubMed; (b) D. L. Hughes in Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer-Verlag, Berlin, 1999, Vol. III pp. 1273–1294, and Supplement 1, 2004, pp. 161–169 Search PubMed.
- (a) P. Melchiorre, Angew. Chem., Int. Ed., 2009, 48, 1360–1363 CrossRef CAS PubMed; (b) A-N. Alba, M. Viciano and R. Rios, ChemCatChem., 2009, 1, 437–439 CrossRef CAS.
- (a) N. Vignola and B. List, J. Am. Chem. Soc., 2004, 126, 450–451 CrossRef CAS PubMed. For a mechanistic insight, see: (b) A. Fu, B. List and W. Thiel, J. Org. Chem., 2006, 71, 320–326 CrossRef CAS PubMed.
- For cascade reactions including intramolecular α-alkylation of aldehydes as a key step, see: (cyclopropanes) (a) J. Lv, J. Zhang, Z. Lin and Y. Wang, Chem.–Eur. J., 2009, 15, 972–979 CrossRef CAS PubMed; (b) R. Rios, H. Sundén, J. Vesely, G.-L. Zhao, P. Dziedzic and A. Córdova, Adv. Synth. Catal., 2007, 349, 1028–1032 CrossRef CAS; (c) H. Xie, L. Zu, H. Li, J. Wang and W. Wang, J. Am. Chem. Soc., 2007, 129, 10886–10894 CrossRef CAS PubMed; (d) V. Terrasson, A. Van der Lee, R. M. De Figueiredo and J. M. Campagne, Chem.–Eur. J., 2010, 16, 7875–7880 CrossRef CAS PubMed (cyclopentanes); (e) I. Ibrahem, G.-L. Zhao, R. Rios, J. Vesely, H. Sundén, P. Dziedzic and A. Córdova, Chem.–Eur. J., 2008, 14, 7867–7879 CrossRef CAS PubMed; (f) R. Rios, J. Vesely, H. Sundén, I. Ibrahem, G.-L. Zhao and A. Córdova, Tetrahedron Lett., 2007, 48, 5835–5839 CrossRef CAS; (g) D. Enders, C. Wang and J. W. Bats, Angew. Chem., Int. Ed., 2008, 47, 7539–7542 CrossRef CAS PubMed.
- Using α-bromo ketones/esters or trifluoromethyl iodide as an alkylating reagent and merging aminocatalysis with photoredox catalysis (Ru2+, Ir+ complexes) (a) D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–79 CrossRef CAS PubMed; (b) D. A. Nagib, M. E. Scott and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 10875–10877 CrossRef CAS PubMed. See, also: (c) A. E. Allen and D. W. C. MacMillan, J. Am. Chem. Soc., 2010, 132, 4986–4987 CrossRef CAS PubMed. α-Carbofunctionalization of aldehydesvia a SOMO activation strategy and reagents other than halides; (d) T. D. Beeson, A. Mastracchio, J.-B. Hong, K. Ashton and D. W. C. MacMillan, Science, 2007, 316, 582–585 CrossRef CAS PubMed; (e) J. M. Um, O. Gutierrez, F. Schoenebeck, K. N. Houk and D. W. C. MacMillan, J. Am. Chem. Soc., 2010, 132, 6001–6005 CrossRef CAS PubMed and references therein. Also see: (f) K. C. Nicolaou, R. Reingruber, D. Sarlah and S. Bräse, J. Am. Chem. Soc., 2009, 131, 2086–2087 CrossRef CAS PubMed (Correction: J. Am. Chem. Soc. 2009, 131, 6640); (g) F. Benfatti, M. G. Capdevila, L. Zoli, E. Benedetto and P. G. Cozzi, Chem. Commun., 2009, 5919–5921 RSC.
- R. R. Shaikh, A. Mazzanti, M. Petrini, G. Bartoli and P. Melchiorre, Angew. Chem., Int. Ed., 2008, 47, 8707–8710 CrossRef CAS PubMed.
- (a) P. G. Cozzi, F. Benfatti and L. Zoli, Angew. Chem., Int. Ed., 2009, 48, 1313–1316 CrossRef CAS PubMed; (b) F. Benfatti, E. Benedetto and P. G. Cozzi, Chem.–Asian J., 2010, 5, 2047–2052 CrossRef CAS PubMed . For a similar approach applied to ketones; (c) L. Zhang, L. Cui, X. Li, J. Li, S. Luo and J.-P. Cheng, Chem.–Eur. J., 2010, 16, 2045–2049 CrossRef CAS PubMed.
- A. R. Brown, W.-H. Kuo and E. N. Jacobsen, J. Am. Chem. Soc., 2010, 132, 9286–9288 CrossRef CAS PubMed.
- For α-allylation of aldehydes using combined metal/organocatalysis, see: (a) (Pd/Brønsted acid asymmetric catalysis) S. Mukherjee and B. List, J. Am. Chem. Soc., 2007, 129, 11336–13337 CrossRef CAS PubMed; (b) (Pd/pyrrolidine catalysis) I. Ibrahem and A. Córdova, Angew. Chem., Int. Ed., 2006, 45, 1952–1956 CrossRef CAS PubMed; (c) (Intramolecular) F. Bihelovic, R. Matovic, B. Volovic and R. N. Saccic, Org. Lett., 2007, 9, 5063–5066 CrossRef CAS PubMed.
- Recent work by Clive and co-workers described a general carbocyclization strategy based on an intramolecular alkylation of carbanions, mainly from active methylenes and CsCO3 or DBU, thetered with an A-type moiety (X = OAc). The study includes an example of enamine-mediated intramolecular aldehyde α-allylation reaction, although the authors conclude the latter approach is clearly inferior and did not establish the ee of the cyclic aldehyde product: L. Wang, B. Prabhudas and D. L. J. Clive, J. Am. Chem. Soc., 2009, 131, 6003–6012 CrossRef CAS PubMed.
- Cinchona alkaloid-catalyzed enantioselective allylation reactions with carbonates of type A (X = OBoc) wherein the control of the stereochemistry relies on the electrophilic component of the reaction (quinuclidine catalyst-containing chiral transient adduct), see: (active methylenes) (a) Y. Du, X. Han and X. Lu, Tetrahedron Lett., 2004, 45, 4967–4971 CrossRef CAS; (b) D. J. V. C. van Steenis, T. Marcelli, M. Lutz, A. L. Spek, J. H. van Maarseveen and H. Hiemstra, Adv. Synth. Catal., 2007, 349, 281–286 CrossRef CAS; (c) H.-L. Cui, J. Peng, X. Feng, W. Du, K. Jiang and Y.-C. Chen, Chem.–Eur. J., 2009, 15, 1574–1577 CrossRef CAS PubMed. (Indoles); (d) H.-L. Cui, J. Peng, J. Lei, K. Jiang and Y.-C. Chen, Angew. Chem., Int. Ed., 2009, 48, 5737–5740 CrossRef CAS PubMed.
- S. Zhu, Y. Wang and D. Ma, Adv. Synth. Catal., 2009, 351, 2563–2566 CrossRef CAS.
- (a) M. Marigo, T. C. Wabnitz, D. Fielenbach and K. A. Jørgensen, Angew. Chem., Int. Ed., 2005, 44, 794–797 CrossRef CAS PubMed; (b) Y. Hayashi, H. Gutoh, T. Hayashi and M. Shoji, Angew. Chem., Int. Ed., 2005, 44, 4212–4215 CrossRef CAS PubMed; (c) See, also: U. Grošelj, D. Seebach, D. M. Badine, W. B. Schweizer, A. K. Beck, I. Krossing, P. Klose, Y. Hayashi and T. Uchimaru, Helv. Chim. Acta, 2009, 92, 1225–1259 CrossRef.
- Isolable ammonium salts from halides of type A and some selected tertiary amines and pyridines have recently been reported: M. Baidya, G. Y. Remennikov, P. Mayer and H. Mayr, Chem.–Eur. J., 2010, 16, 1365–1371 CrossRef CAS PubMed.
- (a) M. Baidya, S. Kobayashi, F. Brotzel, U. Schmidhammer, E. Riedle and H. Mayr, Angew. Chem., Int. Ed., 2007, 46, 6176–6179 CrossRef CAS PubMed; (b) M. Baidya and H. Mayr, Chem. Commun., 2008, 1792–1794 RSC.
- Whereas DMAP led to the α-alkylation product 6 cleanly, with DABCO as the coadyuvant, a small amount (2–5%) of the Stetter adduct was also produced. No product from N–alkylation catalyst was detected in either case. See ESI† for details.
- L. Albrecht, B. Richter, H. Krawczyk and K. A. Jørgensen, J. Org. Chem., 2008, 73, 8337–8343 CrossRef CAS PubMed.
- A. K. Chatterjee, J. P. Morgan, M. Scholl and R. H. Grubbs, J. Am. Chem. Soc., 2000, 122, 3783–3784 CrossRef CAS.
- Aldehyde protection was necessary, otherwise racemisation occurred during RCM.
- See the ESI† for details.
- For kinetic data and a mechanistic discussion on the reaction between stabilized metal carbanions and 2-(halomethyl) vinyl ketones mediated by ammonium salts, see ref. 16.
- For examples of ammonium salts that first direct reaction manifolds while then acting as leaving groups, see: (a) S. France, D. J. Guerin, S. J. Miller and T. Lectka, Chem. Rev., 2003, 103, 2985–3012 CrossRef CAS PubMed; (b) M. J. Gaunt and C. C. Johansson, Chem. Rev., 2007, 107, 5596–5605 CrossRef CAS PubMed.
- Formation of 12via a SN2 process involving 2 and DMAP shows a 20.7 kcal mol−1 energy barrier (see scheme below). Similarly, the subsequent SN2 attack of the enamine on 12 holds a high energy barrier of 24.3 kcal mol−1. See the ESI† for details
.
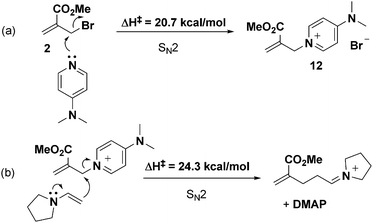
Footnote |
| † Electronic Supplementary Information (ESI) available: Experimental procedures, spectral data for all new compounds, stereochemical proofs, and detailed computational data. See DOI: 10.1039/c0sc00402b |
| This journal is © The Royal Society of Chemistry 2011 |