Exploiting CH-π interactions in supramolecular hydrogels of aromatic carbohydrate amphiphiles†
Louise S.
Birchall
*a,
Sangita
Roy
a,
Vineetha
Jayawarna
a,
Meghan
Hughes
a,
Eleanore
Irvine
a,
Gabriel T.
Okorogheye
b,
Nabil
Saudi
b,
Emiliana
De Santis
b,
Tell
Tuttle
a,
Alison A.
Edwards
*b and
Rein. V.
Ulijn
a
aWestCHEM, Dept of Pure & Applied Chemistry, University of Strathclyde, Glasgow, G1 1XL, UK. E-mail: louise.birchall@strath.ac.uk
bThe Medway School of Pharmacy, The Universities of Kent and Greenwich at Medway, Central Avenue, Chatham Maritime, Kent, ME4 4TB, UK. E-mail: a.a.edwards@kent.ac.uk
First published on 12th May 2011
Abstract
A novel class of supramolecular hydrogels derived from amino sugars is reported, where the self-assembly of aromatic carbohydrate amphiphiles is driven by CH-π interactions, rather than π–π stacking and H-bonding associated with gelators based on aromatic peptide amphiphiles. Spectroscopic data is provided as evidence for this mode of self-assembly and in silico studies revealed that a combination of CH-π and T-stacking of the fluorenyl groups contribute to the formation of the aggregated structures.
1. Introduction
Carbohydrates carry out a myriad of biological functions by acting as energy sources, providing structural rigidity and taking part in binding events involved in biological recognition; this is important to both healthy and disease specific molecular processes. With the introduction of automated solid-phase synthesis of oligosaccharides1 and glycoarrays,2 high throughput screening is now possible for lectin and antibody specificity, carbohydrate binding proteins, enzyme activity and specificity and pathogen identification. Glycopolymers are currently being investigated to exploit these interactions in targeted drug delivery systems.3A relatively new type of interaction has been identified where the apolar patches of carbohydrates bind to aromatic moieties within proteins. Non-covalent forces such as CH-π, van der Waals and hydrophobic effects have been suggested to play a role.4,5 Studies into CH-π interactions have been carried out using a combination of analytical and computational methods.4,6–11 The nature of CH-π and π–π (T-shape) interactions in organic and organometallic compounds as well as bioconjugates has been extensively reviewed.12,13 Inspired by these findings, we sought to exploit CH-π interactions to drive the self-assembly of aromatic carbohydrate amphiphiles in the formation of supramolecular hydrogels.
Supramolecular hydrogels are generated from the self-assembly of gelator molecules through non-covalent interactions such as H-bonding and π–π stacking, which immobilise water within the 3D fibrous network.14–16Gelators possess molecular properties which provide a delicate hydrophobic–hydrophilic balance. There are many examples of peptide-derived hydrogels which are responsive to externally applied stimuli such as changes in solvent polarity, temperature,17 pH,18 ionic strength19 or enzymatic catalysis.20 Such materials are currently being investigated for applications in drug delivery21,22 and cell culture.23,24
A number of carbohydrate-containing hydrogels have been studied. Shinkai and co-workers produced a series of methyl 4,6-O-benzylidene monosaccharides which self-assembled to form fibre networks based on H-bonding interactions. By exploiting the chiral diversity inherent in monosaccharides, they could manipulate gelation properties in various solvents, including water.25 Aromatic carbohydrate amphiphiles have been shown to form both organo- and hydrogels through the formation of an inter-digitated bi-layer structure, where the aqueous gel was found to be stabilized by a combination of H-bonding, π–π interactions and hydrophobic forces.26 Hamachi and co-workers developed a range of thermally responsive supramolecular polymers based on glycosylated amino acid (GalNAc-aa) derivatives.27 One of which was developed into a hydrogel capsule which could respond, by selective digestion, to a prostate specific antigen for the sensing and targeting of prostate cancer.28 In these systems, robust H-bonding networks, assisted by hydrophobic packing, allowed the gel structure to be maintained. Unsaturation of the alkyl chain in aromatic carbohydrate amphiphiles has been shown to reduce the mechanical strength of supramolecular gels formed from organic/aqueous mixtures, where H-bonding and π–π stacking assisted the self-assembly.29 John et al. have described the synthesis of amygdalin-fatty acid hydrogelators which assembled via a combination of H-bonding, π–π stacking and van der Waals interactions. These gels were used for the enzyme triggered delivery of curcumin.30 Aromatic functionalised peptide carbohydrate derivatives (Nap-L/D-Phe-D-glucosamine) form biocompatible hydrogels which have been shown to improve wound healing in mice.31Self-assembly was proposed to be via the formation of a β-sheet like super structure, and a suggested lack of π–π stacking of the Nap groups in the gel phase. A series of carbohydrate-based low molecular weight hydro- and organogelators have been produced from molecules containing methyl 4,6-O-benzylidene-2-deoxy-2-amino-α-D-glucopyranoside as the head group which was functionalised with carbamates and esters,32 and amides and ureas.33 In this case, a combination of hydrophobic and aromatic interactions and H-bonding determined the self-assembly. A carbohydrate derived hydrogel (from methyl 4,6-O-(4′-aldehyde-phenylidene)-α-D-glucopyranoside) has been developed that was not only thermally responsive but also exhibited cysteine and pH responsiveness.34 This was due to the presence of both the aldehyde and acetyl groups in its structure; self-assembly was driven by H-bonding and hydrophobic interactions. Gel-to-solution transitions could be triggered by reaction of the cysteine and the aldehyde to form a thiazolidine derivative, or the dissociation of the acid sensitive acetyl moiety into methyl-α-D-glucopyranoside and tetraphthalaldehyde. Fang and co-workers produced a series of glucose-based naphthalene derivatives with linkers containing hydrazine or ethylenediamine which could gel water through a combination of π–π stacking and intermolecular H-bonding.35 In summary, previously described carbohydrate-containing gelator molecules all assemble via a combination of H-bonding, hydrophobic/hydrophilic and van der Waals interactions, and π–π stacking.
We sought to design simple molecular hydrogelators that contained only an aromatic moiety and a carbohydrate residue, with a view to directing self-assembly under physiological conditions viaCH-π interactions. This was due to the interest in CH-π interactions, the structural and stereo-diversity accessible and the potential applications for biospecific recognition of carbohydrate-based hydrogels. To simplify the synthesis, amino sugars were employed to give increased specificity for the C-2 position over the other free hydroxyl groups present. We chose to derivatise two amino sugars of galactose and glucose stereochemistry, which are epimeric at the C-4 position. Three different aromatic moieties were chosen, two of which have previously been shown to direct the self-assembly of peptide-based hydrogels by providing the π-stacking motif.36–38 We will show that only compounds containing a fluorenylmethoxycarbonyl (Fmoc) moiety will form hydrogels, and that their assembly is not driven by face-to-face π–π stacking and H-bonding interactions as observed with Fmoc-peptide based systems, but is instead driven by a combination of CH-π interactions and T-stacking of fluorenyl groups.
2. Experimental
2.1 Materials and methods
All commercial reagents were used as supplied. N,N-Dimethylformamide (DMF) was purchased dry from the Aldrich chemical company in sure-seal bottles. All other solvents were used as supplied (analytical or HPLC grade), without prior purification. Synthesis of compounds 1–6 was performed under an atmosphere of argon and at room temperature. Thin layer chromatography (tlc) was performed on aluminium sheets coated with 60 F254silica. Sheets were visualised using Hanessian stain (0.2% w/v cerium(IV) sulfate and 5% ammonium molybdate in 2 M sulfuric acid), ninhydrin stain or a UV light source (254/352 nm). Low resolution mass spectra (LRMS, m/z) were recorded on a Finnigan LCQ duo using electrospray (ES) detection. 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on a 500 MHz JEOL FT NMR spectrometer (1H: 500 MHz and 13C: 125 MHz) in the deuterated solvent stated and interpreted with the aid of COSY and HSQC spectra. 13C multiplicities were assigned using a DEPT sequence. All chemical shifts (δ) are quoted in ppm and coupling constants (J) given in Hz. Residual signals from the solvents were used as an internal reference.2.2 Synthesis
Compounds 1–6 (Fig. 1) were all prepared in one step from the hydrochloride salts of D-galactosamine or D-glucosamine (GalNH2·HCl and GlcNH2·HCl) in good yield by the selective reaction of the amino group. Synthesis of compounds 1–6 and characterisation by NMR and LRMS is detailed in the Supplementary Information (SI 1).† | ||
| Fig. 1 Structures of aromatic carbohydrate amphiphiles, containing a range of aromatic moieties and either a galactosamine or glucosamine residue. | ||
2.3 Hydrogel preparation
The aromatic carbohydrate amphiphiles were suspended in either water (1 mL) or phosphate buffered saline (PBS, 1 mL). The suspension was sonicated to aid dissolution. The mixtures were then heated to ∼70–80 °C to form either a clear solution (1–4) or a milky suspension (5–6) which was allowed to cool to room temperature. Hydrogelation was assessed by inversion of the reaction vial.2.4 Transmission electron microscopy (TEM)
Carbon-coated copper grids (No. 400) were glow discharged for 5 s and placed shiny side down on the surface of a 10 μL droplet of sample of hydrogel (formed from compound 1 or 2, 7.5 mM) for less than 5 s. Excess gel was removed by blotting with a filter paper and then 10 μL of negative stain (Nanovan: 1% aqueous methylamine vanadate, obtained from Nanoprobes) was placed on the top of the gel on the grid and allowed to dry for 10 mins. The dried specimens were then imaged using a LEO 912 energy filtering transmission electron microscope operating at 120 kV fitted with a 14 bit/2 K Proscan CCD camera.2.5 Atomic force microscopy (AFM)
Trimmed mica sheets were adhered to AFM support stubs using clear nail varnish. Clean mica surfaces were prepared by removing the top layer of mica with adhesive tape immediately before sample deposition. A sample of hydrogel (0.25 mL) was suspended in water (2.25 mL), and vortexed to ensure thorough mixing. The resulting suspension (50 μL) was deposited onto the mica covered slides, and placed in an incubator (37 °C) overnight. AFM analysis of the samples was carried out on a DPN 5000® nanofabrication system (NanoInk Inc, Skokie, IL) using a close-contact mode cantilever (AppNano, USA). High resolution images were obtained at 0.5 Hz.2.6 Oscillatory rheology
To verify the mechanical properties of the resulting hydrogels, dynamic frequency sweep experiments were carried out on a strain-controlled rheometer (CSL500 Carri-Med Rheometer) using parallel-plate geometry (20 mm diameter). Hydrogels of 1 and 2 were prepared as described in Section 2.3. The experiments were performed at 25 °C and this temperature was controlled throughout the experiment using an integrated electrical heater. Additional precautions were taken to minimize solvent evaporation and to keep the sample hydrated: a solvent trap was used and the internal atmosphere was kept saturated. To ensure the measurements were made in the linear viscoelastic regime, an amplitude sweep was performed and the results showed no variation in elastic modulus (G′) and viscous modulus (G′′) up to a strain of 0.01%. The dynamic modulus of the hydrogel was measured as a frequency function, where the frequency sweeps were carried out between 1 and 40 Hz. The measurements were repeated three times to ensure reproducibility, with the average data shown.2.7 Circular dichroism
Circular dichroism (CD) spectra were measured on a Jasco J600 spectropolarimeter in a 0.1 mm pathlength cylindrical cell, with 1 s integration, step size of 1 nm, a single acquisition and a slit width of 1 nm due to the dynamic nature of the system. The gel was formed within the cell and then heated to 75 °C to obtain a solution to ensure consistency of concentration for spectroscopic measurement.2.8 Fluorescence emission spectroscopy
Fluorescence emission spectra were recorded from 300–600 nm, exciting at either 295 nm (for compounds 1–4) or 272 nm (for compounds 5 and 6) using slit settings of 3 nm, on a Jasco FP-6500 spectrofluorimeter with an attached Julabo F25 waterbath at either 75 or 20 °C. Samples were allowed to equilibrate in the fluorimeter for 5 min prior to taking the measurement. Experimental data was acquired in triplicate and the average data shown.2.9 Computational methodology
In order to investigate the underlying structural motif that could result in the self-assembly of the fluorenyl derivatised amino sugars, the structures of four representative systems (1–4) were determined. For each compound, the initial geometries, in both the α-pyranose and β-pyranose anomeric forms, were generated and subjected to a conformational search using the molecular mechanics forcefield, MM3, as implemented in Spartan.39 This process generated 100 starting conformations of each structure which differed, to varying degrees, by rotations of the rotatable bonds. Each of the conformers, generated through the conformational search, were then optimized at the OM3-D40–43 semi-empirical level of theory (within the MNDO0444 program) in order to generate an energy based ordering of the conformers. The OM3-D optimizations resulted in distinct clusters of conformers, based on their energy.For each pyranose anomer of the four compounds, the Boltzmann weighting of the OM3-D optimized conformers was determined and those conformers that constituted ∼99% of the probable structures (typically 4–13 of the lowest energy conformers) were re-optimized at the RI45-BLYP46,47-D48/def2-TZVP49 level of theory (dispersion augmented density functional theory) as implemented in TurboMole.50 The re-optimization at the density functional level of theory resulted in distinct clusters (up to 5) for each set of low energy conformers of a molecule. Of these, the lowest energy optimized conformer, in the most stable anomeric form, for each compound (Fig. 2) was used to investigate the relative binding energies of various dimer configurations (vida infra). Each of the dimer configurations were subjected to a geometry optimization using the RI-BLYP-D/def2-TZVP level of theory.
 | ||
| Fig. 2 Optimised structures of the lowest energy conformers for the α-pyranose anomer of 1 and the β-pyranose anomers of 2–4. | ||
3. Results and discussion
3.1 Synthesis
For the synthesis of compounds 1–6 (Fig. 1), after removal of the reaction solvent, the poor solubility of the products facilitated their isolation by precipitation. This enabled impurities to be removed by simple washing of the precipitate with water and dichloromethane (DCM). The formation of the Fmoc derivatives (1 and 2) was achieved by treatment of an aqueous solution of the amino sugar (GalNH2·HCl and GlcNH2·HCl, respectively) with sodium hydrogen carbonate followed by drop wise addition of a solution of Fmoc-Cl in dioxane. GalNHFmoc 1 and GlcNHFmoc 2 were isolated as precipitates in 82% and 93% yield, respectively. Compounds 3–6 were prepared by peptide coupling with TBTU between the appropriate amino sugar and aromatic acid. N,N-Diisopropylethylamine (DIPEA) was added to a solution of the amino sugar in DMF followed by addition of O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate (TBTU) and either 9-fluorenylacetic acid or 2-naphthylacetic acid. The desired products were isolated viaprecipitation in unoptimised yields of 63, 76, 32 and 60% for 3–6 respectively. It is noted that GlcNHNap was previously synthesised and tested for hydrogelation by the Xu group.31 Overall, the use of the amino functional group on a monosaccharide scaffold was found to be an efficient way to regiospecifically introduce a range of aromatic moieties in an efficient one step synthesis.3.2 The gelation ability of compounds 1–6
The ability of compounds 1–6 to form hydrogels was assessed by suspending each compound in water and then increasing the temperature to ∼70–80 °C. Cooling the resulting solution to room temperature led to formation of translucent hydrogels from 1 and 2, however, compounds 3–6 failed to form supramolecular hydrogels (Table 1). Compounds 3 and 4 dissolved at high temperatures but formed a precipitate upon cooling. Compounds 5 and 6 did not dissolve upon heating (even at 90 °C) and failed to form a hydrogel. This was previously observed by Xu et al. for compound 6.31| Compound number | Concentration in water (mM) | Appearance | cLogP |
|---|---|---|---|
| 1 | 5, 7.5 | Self-supporting hydrogel | 2.02 |
| 2 | 5, 7.5, 10 | Self-supporting hydrogel | 2.02 |
| 3 | 5.2, 10.4 | Precipitate | 1.75 |
| 4 | 5.2, 10.4 | Precipitate | 1.75 |
| 5 | 5.8, 8.6 | Precipitate | 1.03 |
| 6 | 5.8, 11.5 | Precipitate | 1.03 |
The pH values for hydrogels 1 and 2 were 7.2 and 7.0, respectively. Compounds 1 and 2 also formed hydrogels in PBS (pH 7.4, 0.01 M phosphate buffer, 0.0027 M potassium chloride and 0.137 M sodium chloride). Hydrogels formed in both water and PBS remained stable during repeated heating and cooling cycles.
The thermally reversible gel-to-sol transition temperature was determined by the vial inversion method (full rheological properties were also determined, vide infra). The Tgel for hydrogels formed from 1 and 2 had values of 46–49 °C and 47–50 °C, respectively. Compounds 1 and 2 had the highest cLogP values, indicating that hydrophobicity plays a key role in aggregation (Table 1).
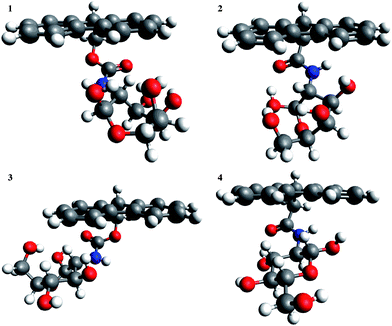
3.3 TEM and AFM
To study the structure of hydrogels 1 and 2, TEM images were obtained (Fig. 3a and b). Both gels revealed bundles of twisted fibres with diameters of 10–24 nm for 1 and 13–25 nm for 2. In both cases, these fibres were in the same range as those observed by Xu et al. for Nap-L/D-Phe-D-glucosamine (in the range of 25–55 nm), and similar in size to those observed for Fmoc-peptide based hydrogels.37AFM images were also obtained and these similarly revealed the fibrous nature of the hydrogels (Supplementary Information, SI 2†).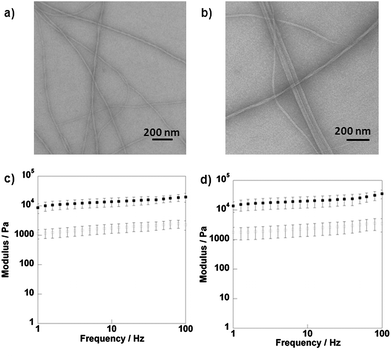 | ||
Fig. 3
TEM images of hydrogels 1 (a) and 2 (b) and rheology data for hydrogels formed from compounds 1 (c) and 2 (d) in water. G′ (■) and G′′ ( ). ). | ||
3.4 Oscillatory rheology
Rheology is used to determine the mechanical properties of supramolecular hydrogels.51Rheology data (Fig. 3c and d) showed that for the hydrogels of 1 and 2, the storage modulus (G′) exceeds the loss modulus (G′′) by an order of magnitude, indicating that they are viscoelastic in nature and behave as typical hydrogels. The hydrogel formed from glucose derivative 2 was mechanically stiffer than that formed from corresponding galactose derivative 1, with G′ values of 21 kPa and 13 kPa, respectively. Both gels had larger G′ values than those reported by Xu et al. for Nap-L/D-Phe-D-glucosamine.31 Similar results were gained for hydrogels formed from compounds 1 and 2 using PBS (Supplementary Information, SI 3†).3.5 Circular dichroism (CD)
Fig. 4a and b show the CD spectra of the Fmoc-carbohydrate hydrogels. In the gel phase, significant CD signals are observed but no CD signal was observed for the compounds in solution state (i.e. high temperature or low concentration). Therefore, the chirality originates from the preferred handedness of the supramolecular assembly, as Fmoc is itself achiral. A distinct peak was observed at 304 nm for both 1 and 2 in the gel state, which corresponds to transitions of the Fmoc chromophores. The sign of the CD signal is the same for both systems, implying a similar chiral arrangement giving rise to a single-handed structure. The observed CD signal is opposite in sign to that observed for a number of previously studied Fmoc-peptide systems.37,52 The level of chiral organization in the self-assembled state is found to be affected by the molecular structure. The glucose derivative 2 showed a more intense 304 nm peak, compared to galactose derivative 1, as a result of more extensive organisation. This is supported by the gel strength measurement, determined through rheology. Furthermore, we have also observed a CD signal at ∼270 nm for these hydrogels, which was also attributed to transitions of the aromatic chromophore. | ||
Fig. 4 CD data for compounds 1 (a) and 2 (b) and fluorescence emission spectra for compounds 1 (c) and 2 (d);  7.5 mM 20 °C, 7.5 mM 20 °C,  7.5 mM 75 °C, 7.5 mM 75 °C,  2.5 mM 20 °C. Data from fluorescence emission spectra of compound 1 (e) and 2 (f) at various concentrations; maximum emission wavelength at 75 °C, maximum emission wavelength at 20 °C, 2.5 mM 20 °C. Data from fluorescence emission spectra of compound 1 (e) and 2 (f) at various concentrations; maximum emission wavelength at 75 °C, maximum emission wavelength at 20 °C,  maximum intensity at 75 °C and maximum intensity at 75 °C and  maximum intensity at 20 °C. maximum intensity at 20 °C. | ||
3.6 Fluorescence emission spectra
Fluorescence spectroscopy provides information about the fluorenyl environment. It was monitored to assess the effect of the local environment on emission wavelength and intensity. The emission spectra of compounds 1 and 2 (0.05 mM, i.e. significantly below the critical gelation concentration) in solvents with varying polarity, revealed that a decrease in solvent polarity resulted in a decrease in fluorescence intensity (Supplementary Information, SI 4.1†).The emission spectra of compounds 1 and 2 (0.01 to 7.5 mM in water) at 75 °C and 20 °C were also recorded; examples of fluorescence emission spectra are shown in Fig. 4c and d and data for all concentrations in Fig. 4e and f. For concentrations of 1 above 0.1 mM, and above 0.75 mM for 2, the maximum intensity decreased with increasing concentration. This indicated that the molecules were experiencing an increasingly hydrophobic environment, i.e. forming aggregates. The maximum wavelength of emission increased with concentration, suggesting a culmination of interactions between the molecules. At concentrations above 3.5 mM for 1 and 4.5 mM for 2 (i.e. around the critical gelation concentration of 4 mM for 1 and 5 mM for 2), there was a slight red shift observed upon gelation (i.e. at 20 °C). For concentrations below 3.5 mM, a blue shift was observed. There was no clear transition in fluorescence intensity observed upon gelation.
These observations were in stark contrast to Fmoc-peptide based hydrogels, where more substantial red shifts (up to 150 nm) in emission wavelength have been observed.37 These shifts were attributed to excimer formation i.e. π–π stacking interactions (Supplementary Information, SI 4.2†). Xu and co-workers observed small red shifts (3–7 nm) for their Nap-L/D-Phe-D-glucosamine hydrogels, which was attributed to insufficient π–π stacking of the Nap residues.31 Molecular modelling (vide infra) suggests that these small red shifts observed upon gelation imply some degree of π–π stacking interactions. Edge stacking is different from the face-to-face stacking observed in a number of Fmoc-peptide assemblies which are characterised by a more extensive red-shift and broad peak at ∼450 nm, indicating long range interactions of stacked fluorenyl groups. These observations provide evidence that self-assembly is not primarily driven by π–π stacking interactions, and is in fact directed by CH-π interactions. The spectra for non-assembling compounds 3–6 showed no significant change in maximum emission wavelength at both 75 °C and at room temperature, however some degree of aromatic interaction was observed (Supplementary Information, SI 4.3†).
3.7 Molecular modelling
While 1 and 2 form gels (indicating a degree of molecular order/self-assembly in solution) the molecules that lack the carbamate linkage to the fluorenyl group, i.e., 3 and 4, do not. Comparison of the lowest energy conformers (Fig. 2) for each structure reveals distinct similarities between the two classes. 1 and 2 both adopt conformations that result in the sugar being oriented towards one side of the fluorenyl group (i.e., in Fig. 2, to the right for 1 and to the left for 2). Conversely, the lowest energy conformers of 3 and 4 both have the sugar centrally located with respect to the fluorenyl group. These structural properties of the monomers have a definite effect on how the units are able to interact to form aggregate structures.In order to investigate this effect, we envisioned four possible modes by which dimers of the monomer units may be formed (Fig. 5a). These include (i) via “J-stacking” which involves a mixture of XH-π interactions between the sugar and the aromatic moiety and T-stacking between the aromatic residues, (ii) aromatic–aromatic π–π stacking (F2F), (iii) H-bonding between the sugars (S2S), and (iv) solely XH-π interactions between the aromatic group and the sugar (F2S).
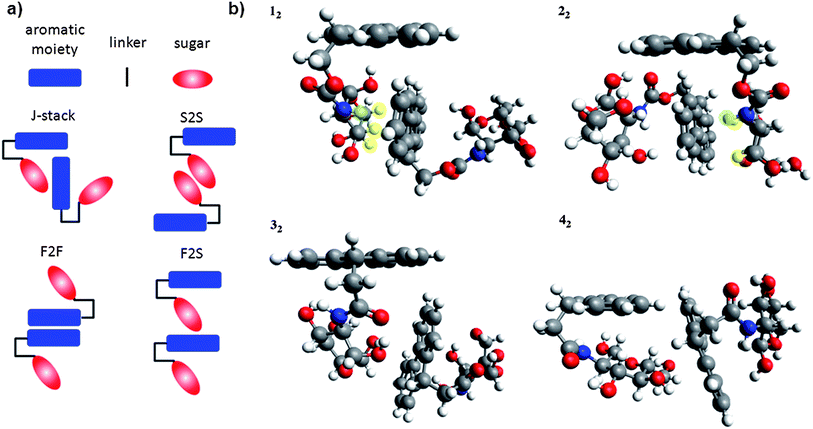 | ||
| Fig. 5 Dimer configurations (a) and RI-BLYP-D optimised geometries of J-stacked dimers (b) of 1–4. | ||
Each of the four possible configurations were optimized at the DFT-D level of theory for the four lowest energy structures of 1–4 and the most stable dimer structures were determined from the relative binding energy of each pair. For the dimers of both 1 and 2, the J-stack orientation (Fig. 5b) resulted in the most favourable binding energy (−21.5 kcal mol−1 and −15.5 kcal mol−1, respectively). The S2S and F2F binding energies of 1 and 2 were ca. −10 kcal mol−1, while the F2S starting geometry reoriented during the optimization to form a J-stacked structure. Critically, the J-stacked motif allows for a consecutive binding of monomers in this structure to form larger aggregates leading to self-assembly. On the other hand, the alternative dimer motifs will not lead to self-assembly as the F2F motif cannot extend past a dimer orientation and the F2S and S2S binding modes predominantly involve H-bonding moieties that will be better satisfied through general solvation of the monomer in the bulk solvent. This is born out in the case of 3 and 4 where the centralised position of the sugar moiety beneath the Fmoc group precludes the possibility of the monomers forming a strong J-stacking interaction (Fig. 5b). In both 3 and 4, the binding energy for the J-stacked motif (−9.5 kcal mol−1 and −7.1 kcal mol−1) is less than the binding energy associated with F2F or S2S motifs, which is again ca. −10 kcal mol−1.
The ability of the Fmoc sugars to self-assemble through a combination of sugar–aromatic and T-stacked dispersion interactions (J-stacking) is necessary for the structures to self-assemble into aggregate structures. It has previously been described that at least three H-atoms of a sugar moiety must be located in the same molecular region, and must be in proximity to the aromatic compound for CH-π stacking to occur.9 We have found that at least two CH's fit these criteria (highlighted in yellow, Fig. 5b) in compounds 1 and 2. There is also a notable contribution from the NH's (highlighted in green, Fig. 5b) and also edge-to-face T-stacking stacking of the fluorenyl groups. Without these combined interactions, competitive H-bonding with the solvent and the terminal nature of the F2F π–π stacking mode eliminates the ability to form aggregates. Thus, the introduction of the ester group into the linker region of the molecule creates the necessary directionality in the compound that orients the sugar molecule to one side of the Fmoc group and allows the J-stacking to occur. Without the ester linkage, the compounds are unable to form stable J-stacked structures and therefore do not self-assemble.
4. Conclusions
In summary, we have produced two novel hydrogelators that represent the first examples of molecular self-assembly which has been driven by a combination of CH-π and T-stacking interactions. Given the variety of carbohydrate molecules accessible, we believe that this novel mode of self-assembly can be used to produce biomaterials where functionality can be tailored towards a range of applications, for example cell culture scaffolds or phase selective gelators for the containment and treatment of oil spills.53Acknowledgements
We thank BBSRC, HFSP, the Leverhulme Trust, ERC and EPSRC for funding, Laurence Tetley for TEM images, Lynsey Aitken for LRMS and Nadeem Javid for assistance with CD. TT thanks the Glasgow Centre for Physical Organic Chemistry for support. AAE gratefully acknowledges the use of NMR facilities at the School of Science, University of Greenwich.References
- O. J. Plante, E. R. Palmacci and P. H. Seeberger, Science, 2001, 291, 1523–1527 CrossRef CAS.
- N. Laurent, J. Voglmeir and S. L. Flitsch, Chem. Commun., 2008, 4400–4412 RSC.
- S. G. Spain and N. R. Cameron, Polym. Chem., 2011, 2, 60–68 RSC.
- J. C. Morales, J. J. Reina, I. Diaz, A. Avino, P. M. Nieto and R. Eritja, Chem.–Eur. J., 2008, 14, 7828–7835 CrossRef CAS.
- C. Tatko, Nat. Chem. Biol., 2008, 4, 586–587 CrossRef CAS.
- J. Screen, E. C. Stanca-Kaposta, D. P. Gamblin, B. Liu, N. A. Macleod, L. C. Snoek, B. G. Davis and J. P. Simons, Angew. Chem., Int. Ed., 2007, 46, 3644–3648 CrossRef CAS.
- E. C. Stanca-Kaposta, D. P. Gamblin, J. Screen, B. Liu, L. C. Snoek, B. G. Davis and J. P. Simons, Phys. Chem. Chem. Phys., 2007, 9, 4444–4451 RSC.
- Z. R. Laughrey, S. E. Kiehna, A. J. Riemen and M. L. Waters, J. Am. Chem. Soc., 2008, 130, 14625–14633 CrossRef CAS.
- K. Ramirez-Gualito, R. Alonso-Rios, B. Quiroz-Garcia, A. Rojas-Aguilar, D. Diaz, J. Jimenez-Barbero and G. Cuevas, J. Am. Chem. Soc., 2009, 131, 18129–18138 CrossRef.
- R. Carrillo, M. Lopez-Rodriguez, V. S. Martin and T. Martin, Angew. Chem., Int. Ed., 2009, 48, 7803–7808 CrossRef CAS.
- M. J. Plevin, D. L. Bryce and J. Boisbouvier, Nat. Chem., 2010, 2, 466–471 CrossRef CAS.
- O. Takahashi, Y. Kohno and M. Nishio, Chem. Rev., 2010, 110, 6049–6076 CrossRef CAS.
- M. Nishio, Y. Umezawa, K. Honda, S. Tsuboyama and H. Suezawa, CrystEngComm, 2009, 11, 1757–1788 RSC.
- R. G. Weiss and P. Terech, ed., Molecular Gels: Materials with Self-Assembled and Fibrillar Networks, Springer, Dordrecht, 2006 Search PubMed.
- J. H. van Esch, Langmuir, 2009, 25, 8392–8394 CrossRef CAS.
- R. V. Ulijn and D. N. Woolfson, Chem. Soc. Rev., 2010, 39, 3349–3350 RSC.
- D. J. Pochan, J. P. Schneider, J. Kretsinger, B. Ozbas, K. Rajagopal and L. Haines, J. Am. Chem. Soc., 2003, 125, 11802–11803 CrossRef CAS.
- J. P. Schneider, D. J. Pochan, B. Ozbas, K. Rajagopal, L. Pakstis and J. Kretsinger, J. Am. Chem. Soc., 2002, 124, 15030–15037 CrossRef CAS.
- B. Ozbas, J. Kretsinger, K. Rajagopal, J. P. Schneider and D. J. Pochan, Macromolecules, 2004, 37, 7331–7337 CrossRef CAS.
- Z. Yang, G. Liang and B. Xu, Acc. Chem. Res., 2008, 41, 315–326 CrossRef CAS.
- Y. Gao, Y. Kuang, Z. F. Guo, Z. H. Guo, I. J. Krauss and B. Xu, J. Am. Chem. Soc., 2009, 131, 13576–13577 CrossRef CAS.
- G. Liang, Z. Yang, R. Zhang, L. Li, Y. Fan, Y. Kuang, Y. Gao, T. Wang, W. W. Lu and B. Xu, Langmuir, 2009, 25, 8419–8422 CrossRef CAS.
- M. Zhou, A. M. Smith, A. K. Das, N. W. Hodson, R. F. Collins, R. V. Ulijn and J. E. Gough, Biomaterials, 2009, 30, 2523–2530 CrossRef CAS.
- E. F. Banwell, E. S. Abelardo, D. J. Adams, M. A. Birchall, A. Corrigan, A. M. Donald, M. Kirkland, L. C. Serpell, M. F. Butler and D. N. Woolfson, Nat. Mater., 2009, 8, 596–600 CrossRef CAS.
- O. Gronwald and S. Shinkai, Chem.–Eur. J., 2001, 7, 4328–4334 CrossRef CAS.
- J. H. Jung, G. John, M. Masuda, K. Yoshida, S. Shinkai and T. Shimizu, Langmuir, 2001, 17, 7229–7232 CrossRef CAS.
- S. Kiyonaka, K. Sugiyasu, S. Shinkai and I. Hamachi, J. Am. Chem. Soc., 2002, 124, 10954–10955 CrossRef CAS.
- M. Ikeda, R. Ochi, A. Wada and I. Hamachi, Chem. Sci., 2010, 1, 491–498 RSC.
- G. John, J. H. Jung, M. Masuda and T. Shimizu, Langmuir, 2004, 20, 2060–2065 CrossRef CAS.
- P. K. Vemula, J. Li and G. John, J. Am. Chem. Soc., 2006, 128, 8932–8938 CrossRef.
- Z. M. Yang, G. L. Liang, M. L. Ma, A. S. Abbah, W. W. Lu and B. Xu, Chem. Commun., 2007, 843–845 RSC.
- G. J. Wang, S. Cheuk, H. Yang, N. Goyal, P. V. N. Reddy and B. Hopkinson, Langmuir, 2009, 25, 8696–8705 CrossRef CAS.
- N. Goyal, S. Cheuk and G. J. Wang, Tetrahedron, 2010, 66, 5962–5971 CrossRef CAS.
- Q. Chen, Y. X. Lv, D. Q. Zhang, G. X. Zhang, C. Y. Liu and D. B. Zhu, Langmuir, 2010, 26, 3165–3168 CrossRef CAS.
- N. Yan, G. He, H. L. Zhang, L. P. Ding and Y. Fang, Langmuir, 2010, 26, 5909–5917 CrossRef CAS.
- M. L. Ma, Y. Kuang, Y. Gao, Y. Zhang, P. Gao and B. Xu, J. Am. Chem. Soc., 2010, 132, 2719–2728 CrossRef CAS.
- A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37–41 CrossRef.
- L. Chen, K. Morris, A. Laybourn, D. Elias, M. R. Hicks, A. Rodger, L. Serpell and D. J. Adams, Langmuir, 2010, 26, 5232–5242 CrossRef CAS.
- Wavefunction Inc., Irvine, CA.
- M. Kolb and W. Thiel, J. Comput. Chem., 1993, 14, 775–789 CrossRef CAS.
- M. Scholten, Ph.D. Thesis, Universität Düsseldorf, 2003.
- T. Tuttle and W. Thiel, Phys. Chem. Chem. Phys., 2008, 10, 2159–2166 RSC.
- W. Weber and W. Thiel, Theor. Chem. Acc., 2000, 103, 495–506 CrossRef CAS.
- W. Thiel, Max-Planck-Institut für Kohlenforschung, Mülheim an der Ruhr, Germany, 2004 Search PubMed.
- K. Eichkorn, O. Treutler, H. Ohm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 240, 283–289 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- TURBOMOLE, COSMOlogic G.m.b.H & Co. KG, Leverkusen, Germany, 2008 Search PubMed.
- C. Q. Yan and D. J. Pochan, Chem. Soc. Rev., 2010, 39, 3528–3540 RSC.
- H. X. Xu, A. K. Das, M. Horie, M. S. Shaik, A. M. Smith, Y. Luo, X. F. Lu, R. Collins, S. Y. Liem, A. M. Song, P. L. A. Popelier, M. L. Turner, P. Xiao, I. A. Kinloch and R. V. Ulijn, Nanoscale, 2010, 2, 960–966 RSC.
- S. R. Jadhav, P. K. Vemula, R. Kumar, S. R. Raghavan and G. John, Angew. Chem., Int. Ed., 2010, 49, 7695–7698 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterisation of compounds, AFM, rheology and fluorescence emission spectra. See DOI: 10.1039/c0sc00621a |
| This journal is © The Royal Society of Chemistry 2011 |