One-step synthesis of composite vesicles: Direct polymerization and in situ over-oxidation of thiophene†
Hang
Sun
a,
Jiating
He
a,
Shuangxi
Xing
ab,
Liangfang
Zhu
a,
Yi Jian
Wong
a,
Yawen
Wang
a,
Hongju
Zhai
ac and
Hongyu
Chen
*a
aDivision of Chemistry and Biological Chemistry, Nanyang Technological University, 21 Nanyang Link, Singapore 637371. Web: http://www.ntu.edu.sg/home/hongyuchen/E-mail: hongyuchen@ntu.edu.sg; Fax: (+65) 67911961
bFaculty of Chemistry, Northeast Normal University, Changchun, 130024, China
cInstitute of Condensed State Physics, Jilin Normal University, Siping, 136000, China
First published on 4th August 2011
Abstract
Composite vesicles with embedded Au nanoparticles are directly prepared from thiophene and Au nanoparticles in aqueous solution in the presence of H2O2 and a catalytic amount of FeCl3. The number, shape, and structure of the vesicles thereof can be readily controlled. We provide evidence that these vesicles contain an aqueous phase in their cavities. The direct synthesis offers an economical route to nanoscale containers and also a rare example for the minimal conditions of vesicle formation.
Introduction
Vesicles are probably the smallest and yet most important containers for chemical reactions. They are essentially aqueous pockets enclosed by a self-assembled membrane of amphiphilic molecules. As the outer walls of cells, mitochondria and chloroplasts, vesicles play essential roles in the compartmentalisation of cellular reactants. With the inclusion of underlying cytoskeleton and embedded membrane proteins, these robust composite vesicles carry out a plethora of vital functions.A tremendous amount of work has been devoted to understanding and emulating the construction of biological vesicles.1Vesicles can be directly synthesized by the self-assembly of amphiphilic molecules: their hydrophobic tails squeeze together forming the membrane core, whereas their hydrophilic heads dissolve in water on both sides of the membrane. As such, the molecules are held together in the membrane by van der Waals attraction and hydrophobic interactions. Hence, vesicles of small amphiphiles such as lipids are often soft and dynamic.2 In contrast, polymer vesicles are less dynamic and mechanically more robust owing to their stronger intermolecular interactions.3 The most common polymer vesicles are those of amphiphilic block copolymers, typically consisting of two or three blocks.3,4 However, it has been reported that less regular polymers, such as the hyperbranched and randomly modified copolymers,5 can also form vesicles. In addition, polymer vesicles can be obtained in organic solution from random copolymers exploiting interchain hydrogen bonding.6
While simple vesicles are nice model systems and a starting point for applications such as drug delivery and imaging,7 it is conceivable that rational incorporation of additional components may bring about advanced functions. A promising prospect is to hybridize them with naturally occurring membrane proteins,8 but the incorporation of nanoparticles (NPs) has also attracted much interest.1b,9 It is of advantage to engineer vesicles particularly robust polymer vesicles with functional properties. For example, magnetic NPs could render vesicles controllable under external field; plasmonic NPs could generate heat upon photoirradiation (e.g., in photodynamic therapy); and fluorescent or SERS-active (surface-enhanced Raman scattering) NPs could offer strong and multiplexed labeling of vesicles.10
To the best of our knowledge, none of the aqueous vesicles in the literature were directly synthesized. Given the complex nature of vesicle self-assembly, polymers were typically prefabricated before being used to make vesicles. In our study of core-shell nanostructures,11,12 we came across an unusual case of composite polymer vesicles (Fig. 1) that were derived from a single monomer, thiophene, in a one-pot reaction. The direct synthesis in water suggested the formation of aqueous vesicles. We were intrigued by this phenomenon, as the direct formation of aqueous vesicles from simple molecules is of relevance to the origin of biological vesicles: primordial life forms needed a membrane boundary before the emergence of complete synthetic apparatus for lipid molecules.
![(a) Schematic illustrations of the Au@PTh core-shell NPs and the Au@vesicle NPs. TEM images of (b) the Au@PTh NPs, [SDS] = 5.0 mM, t = 12 h and (c) Au@vesicle NPs, [SDS] = 2.5 mM, t = 12 h.](/image/article/2011/SC/c1sc00267h/c1sc00267h-f1.gif) | ||
| Fig. 1 (a) Schematic illustrations of the Au@PTh core-shell NPs and the Au@vesicle NPs. TEM images of (b) the Au@PTh NPs, [SDS] = 5.0 mM, t = 12 h and (c) Au@vesicle NPs, [SDS] = 2.5 mM, t = 12 h. | ||
Therefore, in this report, we focused our study on the formation of aqueous vesicles. While the nature of the liquid inside the vesicles cannot be directly probed using existing methods, we carefully analyzed the vesicle growth under various conditions. Converging evidences supported the presence of an aqueous cavity therein. On the basis of our observations, we further proposed that the surface polymerization of thiophene to polythiophene (PTh) on AuNPs and their simultaneous over-oxidation to thiophene sulfone subunits may have given rise to polymers with both hydrophobic and hydrophilic subunits. The phase-segregation of this amphiphilic polymer led to the formation of vesicles.
Results and discussion
Core-shell nanostructure with a AuNP embedded in a PTh nanodroplet (Au@PTh) can be easily synthesized by oxidation of thiophene in water in the presence of AuNPs and sodium dodecylsulfate (SDS): 40 nm citrate-stabilized AuNPs were mixed with 5.0 mM SDS (below its critical micelle concentration (CMC) of 8 mM) and 50 mM thiophene; excess H2O2 (60 mM) and catalytic amount of FeCl3 (9 μM) were then added to initiate the Fe3+-catalyzed oxidative polymerization.13a The reaction mixture was vortexed before it was incubated at 70 °C for 12 h. The colour of the solution turned from red (the colour of AuNPs) to dark brown, indicating the formation of PTh (Fig. 2b). The reaction did not proceed at room temperature (no colour change). This synthesis of Au@PTh is similar to our previous syntheses of core-shell NPs involving polyaniline (PANI)12a and polypyrrole (PPy).12b In these systems, (NH4)2S2O8 was used as the oxidant whereas the current system used H2O2/FeCl3. The surfactant SDS was known to play an important role in stabilizing the AuNPs and their shells during the surface-templated polymerization.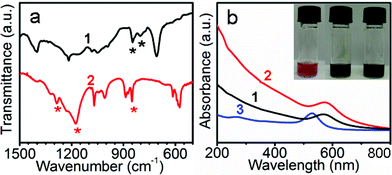 | ||
| Fig. 2 (a) IR spectra of (1) Au@PTh NPs and (2) Au@vesicle NPs. Asterisks indicate peaks of interest. (b) UV-Vis spectra of 1, 2, and (3) citrate-stabilized AuNPs (inset: the photographs of reaction mixtures before heating (left), and after the formation of Au@PTh NPs (middle) and Au@vesicle NPs (right)). | ||
The Au@PTh NPs were then isolated by centrifugation to remove the excess reactants and empty polymer NPs, before they were redispersed in 5.0 mM aqueous SDS. Fig. 1b shows the transmission electron microscopy (TEM) image of typical Au@PTh with 30 nm shell, which appears white against the stained background. The IR spectrum of the purified Au@PTh NPs (Fig. 2a, line 1) shows the typical PTh bands at 800 and 848 cm−1, which were known to arise from the C–H out-of-plane bending of thiophene subunits resulted from α,α-polymerization.13b,13c,14 The surface plasmon absorption peak of AuNPs surprisingly shifted from the 528 nm of citrate-AuNPs to the 566 nm of the Au@PTh NPs (Fig. 2b). This shift was significantly larger than those observed in Au@PANI and Ag@PPy.12a It was likely due to the higher refractive index of PTh15 than those of PANI and PPy.
However, when the [SDS] decreased from 5.0 to 2.5 mM while keeping other conditions unchanged, the morphology of the resulting composite NPs changed dramatically. Fig. 1c shows the TEM image of the vesicle-like structures, with one 40 nm AuNP embedded in the side wall of each nanocomposite (Au@vesicle). With the use of negative stain in TEM imaging, the polymer wall showed a clear contrast against the hollow cavity as well as the stained background. Atomic force microscopy (AFM) images further confirmed that the nanocomposites were not toroids; they were spheroidal with a typical diameter of 270 nm and height of 220 nm.16
The number of vesicular cavities in one nanocomposite is highly correlated with the size of the seed NPs. Using smaller 18 nm AuNP seeds, most of the resulting Au@vesicle NPs (98%) had only one cavity, whereas those with 80 nm AuNPs typically had more than 5 cavities (Fig. 3a–b). The increase in the number of cavities on the larger AuNP seeds was probably due to the inefficient long-range equilibration of the polymer structure at the early stage of vesicle formation (vide infra). Different seeds such as Au nanorods and AgNPs can also be easily incorporated in the PTh-based vesicles using slightly modified methods.16 In general, the Au seeds gave better results than the Ag seeds, which can be partially etched by the H2O2/FeCl3. In all of these examples, most of the nano-composites contained one seed NP. In the few exceptions observed, it appeared that the NPs had aggregated before or during the polymer formation.
![TEM images of (a, b) Au@vesicle NPs with embedded AuNPs of different sizes, dAu = 18 nm and 80 nm, respectively; (c, d) intermediate nanocomposites formed before those in Fig. 1c, t = 1.5 h and 2.5 h, respectively; (e) Au@vesicle NPs with thin walls formed at [thiophene] = 15 mM, [SDS] = 2.5 mM; (f) large Au@vesicle NPs formed at [thiophene] = 50 mM, [SDS] = 0.8 mM; (g) Au@vesicle NPs formed in the absence of SDS; (h) polymer vesicles formed in the absence of SDS and AuNPs.](/image/article/2011/SC/c1sc00267h/c1sc00267h-f3.gif) | ||
| Fig. 3 TEM images of (a, b) Au@vesicle NPs with embedded AuNPs of different sizes, dAu = 18 nm and 80 nm, respectively; (c, d) intermediate nanocomposites formed before those in Fig. 1c, t = 1.5 h and 2.5 h, respectively; (e) Au@vesicle NPs with thin walls formed at [thiophene] = 15 mM, [SDS] = 2.5 mM; (f) large Au@vesicle NPs formed at [thiophene] = 50 mM, [SDS] = 0.8 mM; (g) Au@vesicle NPs formed in the absence of SDS; (h) polymer vesicles formed in the absence of SDS and AuNPs. | ||
To investigate the unusual direct formation of vesicles, we studied the initial states of the nanocomposite by extracting aliquots from the reaction mixture and then quickly drying them on TEM grids. At the early stage of the reaction (1.5 h), polymer nanodroplet was found to attach to the surface of AuNPs, giving eccentric core-shell structures (Fig. 3c).12c Some small cavities of dark colour can be discerned in the shells. Over a longer time, most of the small cavities seemed to have merged and gave rise to one or two dominant dark cavities (d = 50 nm, 2.5 h, Fig. 3d). These cavities only appeared dark when a negative stain, (NH4)6Mo7O24, was applied to the TEM sample. Since the stain is ionic, this observation is an initial indication that the nanocomposites were aqueous vesicles with hydrophilic inner surface. After 12 h (Fig. 1c), the entire AuNP surface was encapsulated in polymer; the vesicle walls increased to 45 nm in thickness and the cavity reached 150 nm in diameter. However, the cavities appeared white even in the presence of the stain. It appeared that with the thicker walls the stain solution can no longer penetrate into the cavities, which, as a result, deflect fewer electrons (brighter in colour) than the side walls. To confirm this reasoning, we decreased the amount of thiophene used and prepared large vesicles of about 10 nm in wall thickness. In Fig. 3e, stain solution appeared to have reached inside the vesicles and formed a pattern typical of dried solution. In cases where more than one cavity was found in a Au@vesicle NP, they often occurred on one side of the AuNP (Fig. 1c), likely because they originated from the same side of an eccentric shell (Fig. 3c).
It is important to understand the role of SDS since it was a key factor in determining whether the Au@PTh or Au@vesicle NPs can be formed. We noticed that the rate of polymerization as indicated by the rate of colour change was significantly higher at high [SDS] than that at low [SDS], with all other conditions kept the same. The dependence of the polymerization rate on the [SDS] highlighted its roles: possibly the ionic Fe3+ can only reacts with thiophene at the surface of SDS micelles. That is, the reaction cannot occur freely in solution but was limited by the available SDS in the solution. The presence of thiophene likely caused SDS to form micelles below its native CMC. Similarly, SDS could adsorb on the thiophene-functionalized Au surface. Both types of the SDS micelles may have provided interfaces facilitating thiophene polymerization. This explains the faster reaction rate at the higher [SDS].
After centrifugation to remove the Au-embedded nanocomposites, the supernatant was dark brown at 5.0 mM SDS but only lightly coloured at 2.5 mM. At the high [SDS], the excess PTh existed in the dark brown supernatant in the form of empty polymer NPs (free of AuNP), as verified by TEM images.16 When [SDS] was further reduced to 0.8 mM, the resulting Au@vesicle NPs were huge (about 1 μm) and appeared as collapsed sacks under TEM (Fig. 3f). With the slow polymerization at this condition, the Au@vesicle NPs actually accumulated larger volumes of polymer than the Au@PTh NPs did at higher [SDS] with a faster reaction rate. This apparent discrepancy was due to the competition of polymerization on the free SDS micelles versus those with AuNPs. At low [SDS], the limited surfactant mostly attached to the thiophene-functionalized Au surface, concentrating the thiophene polymerization there. On the other hand, at high [SDS], the excess surfactant micelles in the solution also competed in the polymerization, leading to faster overall rate but lesser polymer on each AuNP.
In the absence of SDS, some polymer vesicles were still able to form, despite significant AuNP aggregation (Fig. 3g). Hence, the presence of SDS was not a prerequisite for the vesicle formation. Other than affecting the rate of polymerization, the [SDS] did not seem to be directly involved in the shape control of the polymer structure: The isolated Au@PTh NPs were incubated in 2.5 mM SDS and the Au@vesicle NPs in 5.0 mM SDS. No obvious structural change was observed after prolonged incubation.
When the reaction was carried out at 2.5 mM SDS without AuNPs, a large amount of black polymer precipitate was observed. However, when both SDS and AuNPs were absent, the solution showed a brown colour free of precipitate; polymer vesicles were observed in TEM with a broad size distribution (Fig. 3h). The AuNPs acted as seeds that improved the size uniformity of the resulting composite NPs. Without seeds, the uneven growth of polymer structures led to their precipitation. An exception was that when SDS was also absent, the slow polymerization and over-oxidation (vide infra) limited the growth of polymer and prevented its precipitation.
Now that SDS was ruled out as the key factor in controlling the PTh nanostructures, it is important to understand the alternative reasons. In Figs. 1 and 3, the vesicle walls are highly uniform in thickness. One possible reason is that the polymer is able to equilibrate to evenly distribute the shell materials during its growth. The heating at 70 °C was probably also important in promoting polymer mobility.17 In the literature, the vesicles of amphiphilic block copolymers were able to equilibrate; their uniform walls are limited in thickness by the nature of their coiled hydrophobic blocks.17,18 These blocks normally have a preferred physical length and resist being stretched owing to the associated entropic penalty. In such systems, the wall thickness was not easily variable without changing the polymer. In our system, however, the polymer was generated in situ. Thus, the length of the polymer chains must be able to increase during the polymerization. To check the variability of the wall thickness, the Au@vesicle NPs were isolated and used as seeds in additional growth cycles under otherwise identical conditions. The size of the vesicular cavities increased from the initial 150 nm to 200 nm, accompanied by an increase in wall thickness from 45 to 55 nm.16 After a 3rd growth cycle, the cavities increased to 210 nm and their walls to 65 nm (Fig. 4a). Hence, the wall thickness was indeed variable. Interestingly, after the growth cycles, the middle wall separating two cavities always had the same thickness (i.e., grew at a same rate) as that of the outer walls.
![TEM images of (a) Au@vesicle NPs with thick walls formed after 3 growth cycles, [SDS] = 2.5 mM; (b) Au@vesicle NPs after oxidation by excess H2O2, t = 2 h; (c) sample b after purification and an additional growth cycle. Scale bars: 200 nm. (d) XPS spectra in the S2p region of (1) Au@PTh NPs and (2) Au@vesicle NPs. Asterisk indicates peak of interest.](/image/article/2011/SC/c1sc00267h/c1sc00267h-f4.gif) | ||
| Fig. 4 TEM images of (a) Au@vesicle NPs with thick walls formed after 3 growth cycles, [SDS] = 2.5 mM; (b) Au@vesicle NPs after oxidation by excess H2O2, t = 2 h; (c) sample b after purification and an additional growth cycle. Scale bars: 200 nm. (d) XPS spectra in the S2p region of (1) Au@PTh NPs and (2) Au@vesicle NPs. Asterisk indicates peak of interest. | ||
While we think it is likely for the polymer to equilibrate locally to give rise to the uniform shells, the presence of multiple cavities in one nanocomposite (e.g., Fig. 1c) indicated that the structure has not reached the lowest possible surface to volume ratio (S/V), i.e., a global equilibrium was not attained. In particular, the growth of multiple cavities on the 80 nm AuNPs (Fig. 3b) underscored the significance of kinetic pathways: During the initial growth stage, the small cavities formed on the surface of 80 nm AuNPs were in large number and spatially separated. They obviously were not able to fully equilibrate and merge to form a single large cavity. As the PTh grew longer, the equilibration of polymer became even more difficult, and thus multiple cavities resulted. In contrast, on the limited surface of small 18 nm AuNPs, few initial cavities could form and they should be closely positioned (e.g., see Fig. 3c for 40 nm AuNPs), facilitating structural redistribution, and ultimately forming a single cavity.
In the block copolymer systems, the vesicle is a preferred structure with lowest S/V given the limited wall thickness.1b The polymer self-assembly often gives aqueous vesicles with hydrophilic inner surface. However, in our system we do not have direct evidence for the formation of block copolymers.
The alternative explanation for the formation of vesicular cavities is that they were filled with oily droplets. If polymerization only occurred on the SDS-mediated water/thiophene interface, oily droplets could be preserved inside to form vesicle-like structures. Given the limited ingredients in our system, such oily droplets can only be made of thiophene or its short oligomers. They cannot be filled with non-volatile or solid materials because the large vesicles often collapsed after drying (e.g., Fig. 3f). The increase in cavity size with polymerization (Fig. 3c–d, 1c) suggested that the hypothesized oily substances would have higher affinity for the polymer cavity than for the SDS micelles. In theory, equal polymerization at the surface of those nano-droplets could lead to uniform shells that grew in thickness. However, the middle walls separating two cavities would be immersed in a hydrophobic environment; it is unlikely that they should be exposed to a same rate of polymerization and growth.
To further investigate the possibility of oily substances in the cavities, the Au@vesicle NPs were isolated by centrifugation. After removing the supernatant, the concentrated solution was then used for an additional growth cycle without replenishing thiophene. If the hypothesized oily substances did exist and they have a preference for the polymer cavity, they should remain there after the isolation. The subsequent oxidation should decrease the cavity size while increase the shell thickness. However, no significant structural change was observed and the colour of the solution remained unchanged. Given the significant volume ratio of cavity to polymer wall, the oxidation of the oily substances therein should have caused pronounced colour change in the solution. In a different set of experiments, the isolated Au@vesicle NPs were incubated in THF to dissolve away the hypothesized oily substances, followed by centrifugation to remove the supernatant. The Au@vesicle NPs were found to be stable during this process without loss of polymer. The purified NPs were then reintroduced into aqueous SDS solution and used for an additional growth cycle with replenished thiophene. We expect the hydrophobic cavity to collapse in water after removing the hypothesized oily substances. However, vesicles with unaltered structure were observed except that their cavities and walls increased in size.16
Thus, multiple lines of evidences supported the presence of aqueous phase in the vesicular cavities: (a) the appearance of dark stain inside Au@vesicle NPs in TEM images; (b) the uniform thickness and growth of the vesicle walls including the middle wall separating two cavities; (c) control experiments that ruled out oily substances in the cavities; and (d) the correlation between over-oxidation of PTh (giving hydrophilic functional groups) to the formation of new vesicle cavities (vide infra).
Vesicles of conductive polymers are rare. Recently, polymer vesicles formed with all-conjugated amphiphilic diblock copolymers have been prepared.4 Yet, it is highly unusual that aqueous vesicles can be generated from directoxidation of a single monomer. In particular, PTh vesicles can be generated without SDS (Fig. 3g), indicating the presence of hydrophilic functionalities on their surface. There has to be a mechanism for generating hydrophilic units from the hydrophobic PTh. Furthermore, when the purified Au@vesicle NPs were incubated in aqueous H2O2 solution (1%, in large excess) at 70 °C, the colour of the originally dark brown solution turned lighter. After 12 h, the solution became colourless, suggesting the complete loss of conjugated polymer by oxidation. The TEM image of the intermediates isolated during this process (Fig. 4b, t = 2 h) showed reduction in both cavity size and wall thickness. In addition, the number of cavities appeared to have increased. Clearly, the PTh was oxidized and the product material was removed from the hydrophobic wall.
In addition to the characteristic peak of PTh at 851 cm−1, IR spectrum of the Au@vesicle NPs (Fig. 2a, line 2) showed peaks at 1166 and 1288 cm−1. They can be attributed to the symmetric and asymmetric vibrations of the –SO2 moiety,19 supporting the oxidation of thiophene subunits. X-ray photoelectron spectroscopy (XPS) was used to detect oxidized sulfur atoms by analysis of the binding energy of the S2p3/2 electrons (Fig. 4d). In the Au@PTh NPs, the peak at around 163 eV can be attributed to the sulfur atom in thiophene/PTh.14 In contrast, in the Au@vesicle NPs, an additional peak appeared at 168 eV, which is characteristic of sulfur atoms at a high oxidation state (+6 valent), consistent with the formation of thiophene sulfone.20 The first step oxidation of thiophene to thiophene sulfoxide is known to be slower than the second step oxidation to thiophene sulfone;19a this explains the absence of thiophene sulfoxide in our experiments. While thiophene sulfone is unstable in aqueous solution, poly(thiophene sulfone) (PThO2) has been shown to be stable. Actually, PThO2 oligomers and alternating copolymers of PTh-PThO2 have been prepared.21 It has been suggested that Fe3+ ions could be enriched by electrostatic attraction at the vicinity of the SDS micelles;13a they could have catalyzed the polymerization of thiophene and possibly also the over-oxidation of PTh.
Hence, H2O2 played two competing roles in our system: it oxidized Fe2+ to Fe3+ completing the catalytic cycle; and it also oxidized the thiophene subunits in PTh to thiophene sulfone. At high [SDS], the large thiophene/water interface led to fast rate of polymerization and rapid consumption of H2O2 (originally at 1.2 equivalents relative to thiophene). At low [SDS] with the limited reaction interface, the excess H2O2 was able to over-oxidize the PTh to form vesicles. It was known in the literature that thiophene sulfone subunits can be further oxidized by H2O2 to give SO42− ions via a ring opening reaction that breaks the polymer chains.22 But SO42− ions were not detected at a significant level in the supernatant, as we failed to induce BaSO4 precipitate. However, when the purified Au@vesicle NPs were added to a large excess of H2O2 (Fig. 4b), the absence of thiophene monomer promoted the over-oxidation and most of the PTh was removed from the vesicle wall. SO42− ions were detected in the supernatant by inducing BaSO4 precipitate, indicating the breaking of a significant portion of the polymer chains.
The over-oxidized Au@vesicle NPs in Fig. 4b were isolated and then used in an additional growth cycle. Both the cavity size and wall thickness have increased (Fig. 4c), and a larger number of vesicular cavities were found in each nanocomposite in comparison to those before the over-oxidation of polymer (Fig. 1c). Hence, it confirmed that new cavities were created by over-oxidation, and they were enlarged in the subsequent growth cycle. This correlation of the oxidation of PTh to new cavity formation is a further evidence for the aqueous phase therein. Importantly, the middle walls separating two cavities also shrank and increased in unison with the outer walls. Hence, it is likely that the cavities were formed by some sort of self-assembly process that could redistribute the wall materials evenly as the cavities shrank and expand.
On the basis of the above results and analyses, we conclude that the one-pot polymerization and over-oxidation reactions may have led to both hydrophobic PTh and hydrophilic PThO2 subunits. The phase-segregation of the resulting polymer then led to the vesicle self-assembly. This process may be similar to the formation of vesicles from irregular polymers,5 but it was not clear from the literature how exactly it occurred from the phase segregation of polymers to the formation of vesicles. A possibility not yet ruled out is that the PTh-based polymer may have formed segmented amphiphilic blocks, which allowed it to self-assemble in a fashion similar to that of block copolymers. Even in the random polymerization and over-oxidation, it was not entirely impossible that PThO2 blocks could be “pulled” out from the PTh vesicle wall via site-selective over-oxidation at the polymer/water interface.
Conclusions
We report a rare example of direct formation of aqueous polymer vesicles without careful synthetic control of the polymer. The use of a petroleum chemical for making composite vesicles afford an economic route to nanoscale containers. The composite vesicles can be readily tailored by modifying reaction conditions to vary the materials composition of the seed NPs, the wall thickness of the vesicles, the size of vesicle cavities, and the number of cavities per each nanocomposite.Most importantly, the unique system may provide a rare perspective in the minimal conditions of spontaneous vesicle formation. We have demonstrated the evidences supporting the aqueous phase inside the vesicles. Similar aqueous vesicles may be possible using other monomers or even a mixture of monomers. We hope our discovery would attract interest in this and other similar systems. Clearly, further studies are imperative to elucidate the underlying mechanism and to determine whether the mechanism is of relevance to the formation of primordial vesicles before the emergence of synthetic apparatus for lipids.
Acknowledgements
The authors thank the Ministry of Education (ARC 13/09), and the National Research Foundation (CRP-4-2008-06), Singapore, for financial support.Notes and references
- (a) H. Ringsdorf, B. Schlarb and J. Venzmer, Angew. Chem., Int. Ed., 1988, 27, 113 CrossRef; (b) M. Antonietti and S. Forster, Adv. Mater., 2003, 15, 1323 CrossRef CAS; (c) K. Kita-Tokarczyk, J. Grumelard, T. Haefele and W. Meier, Polymer, 2005, 46, 3540 CrossRef CAS; (d) A. Mecke, C. Dittrich and W. Meier, Soft Matter, 2006, 2, 751 RSC; (e) L. F. Zhang and A. Eisenberg, Science, 1995, 268, 1728 CAS.
- J. C. M. Holthuis and T. P. Levine, Nat. Rev. Mol. Cell Biol., 2005, 6, 209 CrossRef CAS.
- (a) B. M. Discher, Y. Y. Won, D. S. Ege, J. C. M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143 CrossRef CAS; (b) D. E. Discher and A. Eisenberg, Science, 2002, 297, 967 CrossRef CAS.
- (a) U. Scherf, S. Adamczyk, A. Gutacker and N. Koenen, Macromol. Rapid Commun., 2009, 30, 1059 CrossRef CAS; (b) U. Scherf, A. Gutacker and N. Koenen, Acc. Chem. Res., 2008, 41, 1086 CrossRef CAS; (c) J. Kim, I. Y. Song and T. Park, Chem. Commun., 2011, 47, 4697 RSC.
- (a) Z. Hordyjewicz-Baran, L. C. You, B. Smarsly, R. Sigel and H. Schlaad, Macromolecules, 2007, 40, 3901 CrossRef CAS; (b) Y. F. Zhou and D. Y. Yan, Angew. Chem., Int. Ed., 2004, 43, 4896 CrossRef CAS.
- (a) B. S. Li, J. W. Chen, C. F. Zhu, K. K. L. Leung, L. J. Wan, C. L. Bai and B. Z. Tang, Langmuir, 2004, 20, 2515 CrossRef CAS; (b) F. Ilhan, T. H. Galow, M. Gray, G. Clavier and V. M. Rotello, J. Am. Chem. Soc., 2000, 122, 5895 CrossRef CAS.
- (a) P. P. Ghoroghchian, P. R. Frail, K. Susumu, D. Blessington, A. K. Brannan, F. S. Bates, B. Chance, D. A. Hammer and M. J. Therien, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 2922 CrossRef CAS; (b) D. A. Christian, O. B. Garbuzenko, T. Minko and D. E. Discher, Macromol. Rapid Commun., 2010, 31, 135 CAS; (c) S. P. Gandhi and C. F. Stevens, Nature, 2003, 423, 607 CrossRef CAS; (d) X. Guo and F. C. Szoka, Acc. Chem. Res., 2003, 36, 335 CrossRef CAS.
- W. Meier, C. Nardin and M. Winterhalter, Angew. Chem., Int. Ed., 2000, 39, 4599 CrossRef CAS.
- (a) M. Krack, H. Hohenberg, A. Kornowski, P. Lindner, H. Weller and S. Forster, J. Am. Chem. Soc., 2008, 130, 7315 CrossRef CAS; (b) Y. Mai and A. Eisenberg, J. Am. Chem. Soc., 2010, 132, 10078 CrossRef CAS.
- (a) M. Yang, T. Chen, W. S. Lau, Y. Wang, Q. Tang, Y. H. Yang and H. Chen, Small, 2009, 5, 198 CrossRef CAS; (b) G. Chen, Y. Wang, M. X. Yang, J. Xu, S. J. Goh, M. Pan and H. Y. Chen, J. Am. Chem. Soc., 2010, 132, 3644 CrossRef CAS.
- (a) T. Chen, M. X. Yang, X. J. Wang, L. H. Tan and H. Y. Chen, J. Am. Chem. Soc., 2008, 130, 11858 CrossRef CAS; (b) G. Chen, Y. Wang, L. H. Tan, M. X. Yang, L. S. Tan, Y. Chen and H. Y. Chen, J. Am. Chem. Soc., 2009, 131, 4218 CrossRef CAS.
- (a) S. X. Xing, L. H. Tan, M. X. Yang, M. Pan, Y. B. Lv, Q. H. Tang, Y. H. Yang and H. Y. Chen, J. Mater. Chem., 2009, 19, 3286 RSC; (b) S. X. Xing, L. H. Tan, T. Chen, Y. H. Yang and H. Y. Chen, Chem. Commun., 2009, 1653 RSC; (c) S. X. Xing, Y. H. Feng, Y. Y. Tay, T. Chen, J. Xu, M. Pan, J. T. He, H. H. Hng, Q. Y. Yan and H. Y. Chen, J. Am. Chem. Soc., 2010, 132, 9537 CrossRef CAS.
- (a) S. J. Lee, J. M. Lee, I. W. Cheong, H. Lee and J. H. Kim, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2097 CrossRef CAS; (b) Y. A. Udum, K. Pekmez and A. Yildiz, Synth. Met., 2004, 142, 7 CrossRef CAS; (c) Z. P. Zhang, F. Wang, F. E. Chen and G. Q. Shi, Mater. Lett., 2006, 60, 1039 CrossRef CAS.
- O. Inganas, B. Liedberg, C. R. Wu and H. Wynberg, Synth. Met., 1985, 11, 239 CrossRef.
- (a) S. Z. Cheng, M. J. Graham, F. W. Harris and S. Jin, 2008, US patent 0130111; (b) N. Fukuzaki, T. Higashihara, S. Ando and M. Ueda, Macromolecules, 2010, 43, 1836 CrossRef CAS.
- See Supporting Information for details.
- C. Liu, G. Chen, H. Sun, J. Xu, Y. Feng, Z. Zhang, T. Wu and H. Chen, Small, 2011 DOI:10.1002/smll.201100443.
- (a) L. F. Zhang and A. Eisenberg, Macromolecules, 1996, 29, 8805 CrossRef CAS; (b) A. Halperin, M. Tirrell and T. P. Lodge, Adv. Polym. Sci., 1992, 100, 31 CrossRef CAS.
- (a) S. Otsuki, T. Nonaka, N. Takashima, W. H. Qian, A. Ishihara, T. Imai and T. Kabe, Energy Fuels, 2000, 14, 1232 CrossRef CAS; (b) W. Qi, Y. Z. Wang, W. Li and L. X. Wu, Chem.-Eur. J., 2010, 16, 1068 CAS.
- (a) M. H. Schoenfisch and J. E. Pemberton, J. Am. Chem. Soc., 1998, 120, 4502 CrossRef CAS; (b) H. Sun, W. F. Bu, Y. C. Li, H. L. Li, L. X. Wu, C. Q. Sun, B. Dong, R. F. Doo, L. F. Chi and A. Schaefer, Langmuir, 2008, 24, 4693 CrossRef CAS.
- (a) G. Barbarella, O. Pudova, C. Arbizzani, M. Mastragostino and A. Bongini, J. Org. Chem., 1998, 63, 1742 CrossRef CAS; (b) T. Yamamoto, I. Nurulla, H. Hayashi and H. Koinuma, Synth. Met., 1999, 107, 137 CrossRef CAS.
- D. S. Zhao, Z. M. Sun, F. T. Li, R. Liu and H. D. Shan, Energy Fuels, 2008, 22, 3065 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization and large-area supporting figures. See DOI: 10.1039/c1sc00267h |
| This journal is © The Royal Society of Chemistry 2011 |