Photocatalytic reductive cyclizations of enones: Divergent reactivity of photogenerated radical and radical anion intermediates†
Juana
Du
,
Laura Ruiz
Espelt
,
Ilia A.
Guzei
and
Tehshik P.
Yoon
*
Department of Chemistry, University of Wisconsin–Madison, 1101 University Avenue, Madison, WI 53706, USA. E-mail: tyoon@chem.wisc.edu; Fax: +1 608 265 4534; Tel: +1 608 262 2268
First published on 23rd August 2011
Abstract
Photocatalytic reactions of enones using metal polypyridyl complexes proceed by very different reaction manifolds in the presence of either Lewis or Brønsted acid additives. Previous work from our lab demonstrated that photocatalytic [2 + 2] cycloadditions of enones required the presence of a Lewis acidic co-catalyst, presumably to activate the enone and stabilize the key radical anion intermediate. On the other hand, Brønsted acid activators alter this reactivity and instead promote reductive cyclization reactions of a variety of aryl and aliphatic enonesvia a neutral radical intermediate. These two distinct reactive intermediates give rise to transformations differing in the connectivity, stereochemistry, and oxidation state of their products. In addition, this reductive coupling method introduces a novel approach to the tin-free generation of β-ketoradicals that react with high diastereoselectivity and with the high functional group compatibility typical of radical cyclization reactions.
Over the past several decades, organic chemists have become increasingly cognizant of the synthetic utility of reactions involving free radicals. These highly reactive species often display reactivity that is orthogonal to that of polar reactive intermediates such as carbanions and carbocations and thus enable the synthesis of complex structures that would otherwise be difficult to assemble.1 The generation of free radicals, however, typically requires toxic, reactive, or otherwise unattractive additives such as tin compounds, trialkylboranes, and peroxides. MacMillan,2 Stephenson,3 and Gagné4 have each recently described methods for the tin-free generation of carbon-centered free radicals from reductive dehalogenation of electron-deficient haloalkanes under photocatalytic conditions.5
We recently reported a superficially similar system for the [2 + 2] cycloaddition of aryl enones (e.g., 1) upon irradiation with visible light in the presence of Ru(bpy)3Cl2 (Scheme 1, path A).6 The key intermediate in these cycloadditions is the radical anion (2) formed upon one-electron reduction of the enone substrate in the presence of a Lewis acidic Li+ additive. Importantly, it quickly became clear to us that the nature of this reactive intermediate7 was quite different than that of neutral radicals such as those involved in MacMillan, Stephenson, and Gagné's systems. The presence of both a charge and an unpaired spin in the enone radical anion is evidently required for formation of the cyclobutane product (3), as the cycloaddition pathway seemed discordant with the 5-exo-trig cyclization reactivity that would typically be expected from a neutral radical.
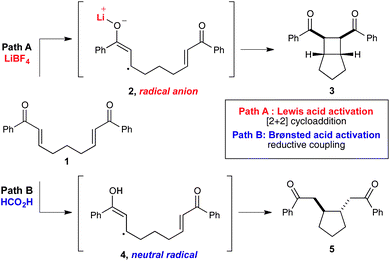 | ||
| Scheme 1 | ||
The realization that these two classes of reactive intermediates might exhibit fundamentally different characteristic reactivity led us to consider how we might access neutral radical intermediates instead of radical anions from photocatalytic reduction of bis(enone) substrates.8–10 In particular, activation of bis(enone) 1 with a Brønsted acid would afford a cationic oxocarbenium species, reduction of which should produce a neutral radical (4, Scheme 1, path B). This intermediate would then be poised to undergo 5-exo-trig cyclization rather than the [2 + 2] cycloaddition preferred by the radical anion.
Thus, we began an investigation of the effect of Brønsted acid additives on the photocatalytic one-electron reduction of bis(enone) 6 (Table 1). The substrate is unchanged in the absence of an acid co-catalyst (entry 1). In the presence of a variety of Brønsted acid additives, however, 6 undergoes reductive cyclization to produce the cyclized diketone 7 (entries 2–6). In all cases, trans-7 is the major product observed, and only traces of products arising from[2 + 2] cycloaddition could be observed. In our survey of Brønsted acids, formic acid showed particular promise for promoting reductive cyclization (entry 4). Further optimization studies revealed that the reaction rate increases in the presence of a larger excess of acid (entry 7), and that lowering the photocatalyst loading to 2.5 mol% produced the desired cycloadduct in 95% yield as a single trans diastereomer (entry 8).
|
|
||
|---|---|---|
| Entry | Conditions | Yieldb |
| a Reactions were irradiated using a 23 W compact fluorescent bulb. b Yields were determined by 1H NMR analysis against an internal standard unless otherwise noted. c Isolated yield. d Other than the light source and reaction time, this entry uses the optimized conditions from ref. 11b. See Supporting Information for experimental details.† | ||
| 1 | 5 mol% Ru(bpy)3Cl2, no acid, 4 equiv i-Pr2NEt | <5% |
| 2 | 5 mol% Ru(bpy)3Cl2, 2 eq H2SO4, 4 eq i-Pr2NEt | <5% |
| 3 | 5 mol% Ru(bpy)3Cl2, 2 eq TFA, 4 eq i-Pr2NEt | 13% |
| 4 | 5 mol% Ru(bpy)3Cl2, 2 eq HCO2H, 4 eq i-Pr2NEt | 58% |
| 5 | 5 mol% Ru(bpy)3Cl2, 2 eq BzOH, 4 eq i-Pr2NEt | 43% |
| 6 | 5 mol% Ru(bpy)3Cl2, 2 eq AcOH, 4 eq i-Pr2NEt | 36% |
| 7 | 5 mol% Ru(bpy)3Cl2, 5 eq HCO2H, 10 eq i-Pr2NEt | 66% |
| 8 | 2.5 mol% Ru(bpy) 3 Cl 2 , 5 eq HCO2H, 10 eq i-Pr2NEt | 95%c |
| 9 | 2.5 mol% DCA, 5 eq HCO2H, 10 eq i-Pr2NEt | <5% |
| 10d | 20 mol% DCA, 60 mol% Ph3P | <5% |
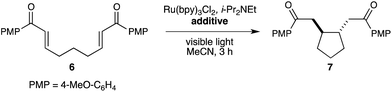
Pandey has described a photocatalytic system for reductive cyclizations of enones that employs 9,10-dicyanoanthracene (DCA) as a photosensitizer along with PPh3 as a co-reductant.11 The optimized conditions for this system required relatively high loadings of the photosensitizer (20 mol%) and the use of a 450 W mercury arc lamp. We have previously argued6 that the longer excited state lifetime (600 ns vs. 15 ns) and broader visible absorption of Ru(bpy)32+ compared to DCA allow lower catalyst loadings and lower-intensity light sources to be used in photocatalytic applications.12,13 Indeed, in comparison experiments using DCA as the photocatalyst, either in place of Ru(bpy)32+ in our optimized protocol (entry 9) or using a 23 W compact fluorescent bulb in place of the 450 W Hanovia lamp in Pandey's protocol (entry 10), we observe no reductive cyclization products after 3 h of irradiation. Thus, the use of Ru(bpy)32+ as a photocatalyst, which results in complete conversion to 7 in the same timeframe, affords a significant improvement over this previous approach to photochemical reductive cyclization.
We next proceeded to examine the scope of reductive cyclization for a series of structurally varied aryl enones under our optimized conditions (Table 2). Symmetrical bis(enone)s bearing electron-donating and -withdrawing substituents are excellent substrates (entries 1–3),14 as are heteroaryl enones (entry 4). Unsymmetrical enones with one aryl component also react smoothly (entries 5 and 6). α-Substituted enoates readily undergo cyclization, although the configuration of the newly formed α-stereocenter is not controlled under these conditions (entry 7). A variety of heteroatomic, substituted, and extended tethering moieties can be utilized (entries 8–11). Furthermore, as is the case with most radical-mediated cyclization reactions, the presence of acidic heteroatomic functionalities on the substrate is not problematic (entries 12 and 13). Unlike tin-mediated radical cyclizations, however, we find that unactivated alkyl bromides are easily tolerated (entry 14), enabling the construction of products containing this useful synthetic handle for further manipulation.
| Entry | Substrate | Product | Time | Yieldc | d.r.d |
|---|---|---|---|---|---|
| a Reactions conducted with Ru(bpy)3Cl2 (2.5 mol%) HCO2H (5 equiv), and i-Pr2NEt (10 equiv) in MeCN and irradiated using a 23 W compact fluorescent bulb. b PMP = 4-MeO-C6H4. c Data represent the averaged isolated yields from two reproducible experiments. d Diastereomer ratios determined by 1H NMR analysis of the unpurified reaction mixtures. | |||||
|
|
||||
| 1 | R1 = R2 = Ph | 2.5 h | 82% | >10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
|
| 2 | R1 = R2 = PMP | 3 h | 95% | >10:1 |
|
| 3 | R1 = R2 = 4-Cl-C6H4 | 1.5 h | 76% | >10:1 |
|
| 4 | R1 = R2 = 2-furyl | 4 h | 69% | >10:1 |
|
| 5 | R1 = PMP, R2 = t-Bu | 6 h | 95% | >10:1 |
|
| 6 | R1 = PMP, R2 = SEt | 9 h | 79% | 10:1 |
|
| 7 |

|
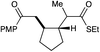
|
12 h | 75% | 1:1 |
|
|
||||
| 8 | X = O | 3.5 h | 93% | >10:1 |
|
| 9 | X = NTs | 4 h | 83% | >10:1 |
|
| 10 | X = CMe2 | 3 h | 73% | >10:1 |
|
| 11 | X = CH2CH2 | 6 h | 92% | >10:1 |
|
|
|
||||
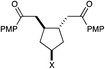
| 12 | X = OH | 5 h | 74% | >10:1 |
|
| 13 | X = NHBoc | 3.5 h | 92% | >10:1 |
|
| 14 |
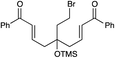
|
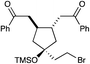
|
3 h | 64% | 10:1 |
We also became interested in exploring the reductive cyclization of aliphatic enones. These compounds are less easily reduced than aryl enones,15 and [2 + 2] cycloadditions were not successful with these substrates under the Li-promoted conditions we previously reported. We were thus pleased to discover that aliphatic bis(enone)s underwent reductive cyclization in high yields, although these substrates required extended reaction times (Table 3, entry 1). We speculated that the use of a more strongly reducing photocatalyst might increase the rate of the cyclization. Indeed, the use of Ir(dtb-bpy)(ppy)2(PF6),16 which MacMillan2b and Stephenson3c have shown to be more effective photocatalysts in reductive dehalogenation processes, enabled the desired cycloadduct to be formed in good yields in only 12 h (Table 3, entry 2).
| Entry | Substrate | Product | Time | Yieldb | d.r.c |
|---|---|---|---|---|---|
|
a Unless otherwise noted, reactions conducted with 2.5 mol% Ir(dtb-bpy)(ppy)2(PF6) in the presence of HCO2H (5 equiv) and i-Pr2NEt (10 equiv) in MeCN. Reactions were irradiated using a 23 W compact fluorescent bulb.
b Data represent the averaged isolated yields from two reproducible experiments.
c Diastereomeric ratios determined by 1H NMR analysis of the unpurified reaction mixtures.
d
Ru(bpy)3Cl2 was used instead of Ir(dtb-bpy)(ppy)2(PF6)2.
e A 2:1 trans:cis mixture of the vinyl nitrile was used for this entry.
|
|||||
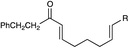
|
|
||||
| 1d | R = COCH2CH2Ph | 36 h | 93% | 5:1 |
|
| 2 | R = COCH2CH2Ph | 12 h | 93% | 5:1 |
|
| 3 | R = CO2Et | 24 h | 77% | >10:1 |
|
| 4e | R = CN | 9 h | 80% | 1:1 |
|
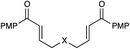
| 5 |
|
|
12 h | 68% | 8:1 |
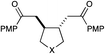
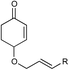
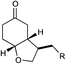
| 6 |
|
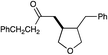
|
12 h | 64% | 2:1 |
|
|
||||
| 7 | R = Ph | 20 h | 60% | 10:1 |
|
| 8 | R = 4-Me-C6H4 | 20 h | 78% | 8:1 |
|
| 9 | R = 4-CF3-C6H4 | 20 h | 91% | 6:1 |
|
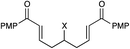
| 10 |
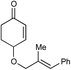
|
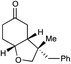
|
24 h | 73% | >10:1 |
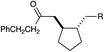
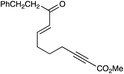
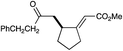
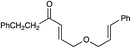
A variety of other Michael acceptors, including acrylate esters (entry 3), vinyl nitriles (entry 4), and alkynoate esters are good reaction partners for reductive coupling with aliphatic enones as well (entry 5). We were particularly interested in examining cyclization reactions with styrenic acceptors. In our previous studies of the lithium-promoted [2 + 2] cycloadditions, we had observed that only Michael acceptors bearing strongly electron-withdrawing groups would react with the photogenerated radical anion intermediates to afford cyclobutane products. However, the neutral radical intermediates we presumed to be involved in these reductive couplings should be able to react with a broader range of less activated alkenes, including styrenic olefins. Indeed, we were delighted to discover that the reductive couplings of linear aliphatic enones with styrenes proceeded in high yield, albeit in lower diastereoselectivity (entry 6).17Cyclizations of cyclic enones with styrenes, on the other hand, proceed with good to excellent diastereoselectivity for a variety of styrenes (entries 7–9), including β-disubstituted styrenes that produce cyclized products containing quaternary carbon stereocenters (entry 10). Reactions involving unactivated aliphatic olefins, however, were not high-yielding.18
From these results, it is clear that the Brønsted acid-mediated radical cyclization is fundamentally different from the Lewis acid-induced radical anion cyclobutanation we previously reported, for several reasons. First, the scope of each reaction is quite distinct; while neither aliphatic enones nor styrenes participate in the radical anion [2 + 2] cycloaddition, they are excellent reaction partners in the radical-mediated reductive cyclization process. Second, the two cyclization events favor divergent stereochemical outcomes; while the [2 + 2] cycloaddition requires a cis ring junction, the reductive cyclizations of enones is generally trans selective. Control experiments preclude the possibility that the reductive cyclization product might arise from initial [2 + 2] cycloaddition followed by reductive scission of the α–α' bond; when the [2 + 2] cycloadduct is independently synthesized and subjected to the conditions for direct reductive cyclization for 2.5 h, the cis reductive cleavage product (8) is formed in only 32% yield (eq 1, Scheme 2). Thus, the cyclobutane is kinetically incompetent to be an intermediate in the reductive cyclization. Moreover, the stereochemistry of the reductive cleavage product is opposite to that observed in the direct reductive cyclization of 1, which further rules out the intermediacy of the cyclobutane in this process.
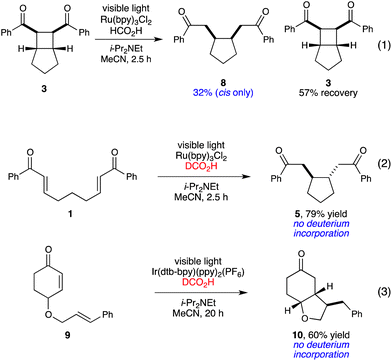 | ||
| Scheme 2 Control experiments. | ||
A third major difference between these processes is the redox balance of the overall transformation. The reductive cyclization constitutes a two-electron reduction of the enone substrate, while the [2 + 2] cycloadditions are net redox-neutral. Under the optimized conditions for reductive cyclization, either the amine base additive or the formate moiety could potentially serve as the terminal reductant for this process. To probe the feasibility of these possibilities, we conducted reductive cyclizations using monodeuterated DCO2H as the Brønsted acid component. These experiments did not result in deuterium incorporation, either under the Ru(bpy)32+ or Ir(dtb-bpy)(ppy)2+ conditions (eq 2 and 3, Scheme 2). Thus, formic acid is presumably serving solely as a Brønsted acid, and the amine base additive, not the formate moiety, is the terminal reductant in this process.19,20
The mechanism outlined in Scheme 3 is consistent with all of the experimental evidence we have collected to date. Visible light irradiation of Ru(bpy)32+ produces a photoexcited state (Ru*(bpy)32+) that can be reductively quenched by i-Pr2NEt to afford an amine radical cation (i-Pr2NEt˙+) and a Ru(bpy)3+ complex. This photogenerated reductant can transfer an electron to the protonated oxocarbenium ion 11 to produce the key neutral radical intermediate 4, which undergoes 5-exo-trig cyclization to yield α-ketoradical 12. Hydrogen atom transfer from the photogenerated amine radical cation, either in a single atom transfer step or by sequential electron- and proton-transfer steps, would then afford the reductive cyclization product 5.
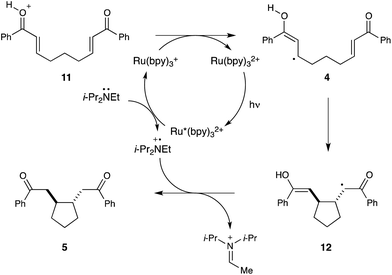 | ||
| Scheme 3 Proposed mechanism for photocatalytic reductive couplings. | ||
Thus, this study underscores the versatility of transition metal polypyridyl photocatalysts in organic synthesis. The same catalysts enable the facile conversion of enones either to radical anions, which participate in an overall redox-neutral [2 + 2] cycloaddition, or to neutral radicals, which result in a net two-electron reductive coupling reaction. The reactivities of these two reactive intermediates are quite distinct from one another,7 as manifested in the different connectivity and oxidation states of their products, their divergent stereochemical outcomes, and the distinct substrate scopes of their transformations, and the appropriate choice of Brønsted or Lewis acidic additive controls the partitioning between these two reaction manifolds.
Conclusions
In summary, we have developed a highly efficient method for the photocatalytic generation of radicals from enones upon irradiation with visible light. The use of a Brønsted acid activator in conjunction with a metal polypyridyl photocatalyst enables the reduction of a variety of aryl and alkyl enones, and the resulting radicals can cyclize with enones and styrenic olefins to produce reductive cyclization products. This represents an attractive method for the initiation of radical reactions that facilitates reactions with the same general reactivity and high functional group compatibility of conventional radical cyclizations without the need for unattractive stoichiometric tin reagents. Furthermore, this study shows that the nature of the reactive intermediate involved in Ru(bpy)32+-catalyzed photochemical transformations can be modulated by the use of either Brønsted or Lewis acid activators. This finding indicates that the scope of reactions that may be accessible via this approach to visible light photocatalysis is remarkably broad. The development of these reactions is an ongoing goal of the research in our lab.Acknowledgements
JD is grateful to the ACS Division of Organic Chemistry for a graduate fellowship. This research was conducted using funds from the ACS PRF (49817-ND1), NIH (GM095666), Sloan Foundation, Beckman Foundation, and Research Corporation. The NMR facilities at UW-Madison are funded by the NSF (CHE-9208463, CHE-9629688) and NIH (RR08389-01).Notes and references
-
(a)
D. I. Davies and M. J. Parrott, Free Radicals in Organic Synthesis; Springer-Verlag: Germany, 1978 Search PubMed
; (b) D. J. Hart, Science, 1984, 223, 883–887 CAS
-
(a) D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS
-
(a) J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756–8757 CrossRef CAS
-
(a) R. S. Andrews, J. J. Becker and M. R. Gagné, Angew. Chem., Int. Ed., 2010, 49, 7274–7276 CrossRef CAS
- For reviews of recent developments in visible light photoredox catalysis, see:
(a) K. Zeitler, Angew. Chem., Int. Ed., 2009, 48, 9785–9789 CrossRef CAS
-
(a) M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886–12887 CrossRef CAS
- For reviews of the reactivity of radical ions, see:
(a) P. I. Dalko, Tetrahedron, 1995, 51, 7579–7653 CrossRef CAS
-
Tin-mediated reductive couplings of enones have previously been reported:
(a) E. J. Enholm and K. S. Kinter, J. Am. Chem. Soc., 1991, 113, 7784–7785 CrossRef CAS
- For Ni-catalyzed cis-selective reductive cyclizations of bis(enone)s, see:
(a) A. V. Savchenko and J. Montgomery, J. Org. Chem., 1996, 61, 1562–1563 CrossRef CAS
- Krische observed reductive cyclization products as components of complex mixtures upon electrochemical reduction of aromatic bis(enone)s:
(a) T.-G. Baik, A. L. Luis, L.-C. Wang and M. J. Krische, J. Am. Chem. Soc., 2001, 123, 6716–6717 CrossRef CAS
-
(a) G. Pandey and S. Hajra, Angew. Chem., Int. Ed. Engl., 1994, 33, 1169–1171 CrossRef
- For an overview of the photophysical properties of Ru(bpy)32+, see: K. Kalyanasundaram, Coord. Chem. Rev., 1982, 46, 159–244 CrossRef CAS
- For the photophysical properties of DCA, see: I. R. Gould, D. Ege, J. E. Moser and S. Farid, J. Am. Chem. Soc., 1990, 112, 4290–4301 CrossRef CAS
- The stereochemistry of the reductive cyclization product of entry 1 was unambiguously established by single-crystal X-ray diffraction. See the Supporting Information for details†.
- H. O. House, L. E. Huber and M. J. Umen, J. Am. Chem. Soc., 1972, 94, 8471–8475 CrossRef CAS
- J. D. Slinker, A. A. Gorodetsky, M. S. Lowry, J. Wang, S. Parker, R. Rohl, S. Bernhard and G. G. Malliaras, J. Am. Chem. Soc., 2004, 126, 2763–2767 CrossRef CAS
- Interestingly, only aliphatic enones cyclize with styrenes; when the styrene is instead tethered to an aryl enone, no productive cyclization is observed using either Ru(bpy)32+ or Ir(dtb-bpy)(ppy)2+. We rationalize this observation by proposing that cyclization occurs via the enol tautomer of the intermediate. The radical produced from the aryl enone would be stabilized by delocalization into the aromatic ring and would thus presumably be less reactive towards cyclization:
. - The major product observed in reactions involving aliphatic olefins resulted from reduction of the enone without productive cyclization.
- This is in contrast to a report by the Stephenson group (ref. 3a), who observed deuterated products in reductive dehalogenation reactions using DCO2H and i-Pr2NEt.
- We observe the formation of acetaldehyde as a by-product, consistent with oxidation of the N-ethyl group of i-Pr2NEt. We cannot rule out the possibility that oxidation of the isopropyl group also occurs; however, the decomposition of trialkylamine radical cations has been studied, and oxidation of the least substituted N-substituent is common. For examples, see:
(a) G. Pandey, P. Y. Reddy and U. T. Bhalerao, Tetrahedron Lett., 1991, 32, 5147–5150 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectral data for all new compounds and crystallographic data for Table 2, entry 1. CCDC reference number 829840. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00357g |
| This journal is © The Royal Society of Chemistry 2011 |