Enzymatic synthesis of 2′-methylseleno-modified RNA†
Vanessa
Siegmund
a,
Tobias
Santner
b,
Ronald
Micura
*b and
Andreas
Marx
*a
aDepartment of Chemistry and Konstanz Research School Chemical Biology, University of Konstanz, Universitätsstraße 10, 78457, Konstanz, Germany. E-mail: andreas.marx@uni-konstanz.de; Fax: +49-7531-885140; Tel: +49-7531-885139
bInstitute of Organic Chemistry and Center for Molecular Biosciences (CMBI), Leopold-Franzens University, Innrain 52a, 6020, Innsbruck, Austria. E-mail: ronald.micura@uibk.ac.at; Fax: +43-512-5072892; Tel: +43-512-5075210
First published on 18th August 2011
Abstract
Selenium-derivatization of RNA is a powerful and advantageous alternative to conventional heavy atom derivatization techniques that are required for the phasing of X-ray crystallographic diffraction data. Among several possibilities, the 2′-methylseleno (2′-SeCH3) modification has been most widely explored and was responsible for a series of important RNA structure determinations, such as the Diels–Alder ribozyme or complexes of antibiotics to HIV dimerization initiation site (DIS) RNA. So far, 2′-SeCH3-RNA has only been accessible by chemical solid-phase synthesis for sizes of up to 50 nucleotides and up to about 100 nucleotides in combination with enzymatic ligation procedures. To overcome this limitation, here we present the enzymatic synthesis of 2′-SeCH3-RNA to open up access for the preparation of long selenium-modified RNA sequences, which cannot be accomplished by conventional chemical synthesis. Therefore, we first elaborated a synthetic route towards the 2′-methylseleno-2′-deoxyribonucleoside triphosphates of cytosine and uridine (2′-SeCH3–CTP and 2′-SeCH3–UTP). With these crucial derivatives in hand, we found that mutants of T7 RNA polymerase are able to incorporate 2′-SeCH3–CMP and 2′-SeCH3–UMP into RNA, while the wild-type polymerase fails to do so. This study demonstrates the efficient enzymatic synthesis of 2′-SeCH3-modified RNA and, thus, provides a thorough foundation for an alternative derivatization strategy in X-ray crystallographic structure analysis of larger RNAs. Such efforts are currently highly requested because of the steadily increasing number of novel non-coding RNAs whose structural features remain to be elucidated.
Introduction
Modified nucleic acids are substantial for the development of new oligonucleotide-based methods and technologies, since they possess altered properties in comparison to their natural counterparts. In addition to the conventional chemical synthesis of modified oligonucleotides on a solid support, the enzymatic synthesis of modified DNA and RNA by nucleic acid polymerases represents an alternative route to overcome the inherent limitations of automated chemical synthesis, such as oligonucleotide size restriction and degree of modification.By using modified 2′-deoxynucleoside triphosphates (dNTPs), DNA polymerases can incorporate modifications into DNA in primer extension reactions and even amplify modified DNA using PCR. Modifications attached to the nucleobase at C5 of pyrimidines or C7 of 7-deazapurines are tolerated by many DNA polymerases.1–9 Therefore, DNA polymerases are widely employed to introduce functionalities into DNA for a variety of applications. Enzymatic synthesis of modified RNA is often accomplished by in vitrotranscription using T7 RNA polymerase in the presence of either modified ribonucleoside triphosphates (NTPs) or of so-called initiator nucleotides to site-specifically modify the 5′-end of the RNA.10–14 When modified NTPs are used for the internal labeling of RNA, the modifications are attached to the nucleobase or at the phosphates.15–18 Nevertheless, modifications at the 2′-position of the ribose are of great interest for further usage of the modified RNAs in siRNA technologies or as aptamers due to their increased stability and resistance towards ribonucleases, their enhanced pharmacokinetic properties and their ability to reduce immunostimulatory effects.19–22 Since wild-type T7 RNA polymerase is not able to efficiently incorporate nucleotides with modifications at the 2′-position into RNA, mutant enzymes have been generated. These T7 RNA polymerase mutants can be used to efficiently incorporate nucleotides with modified 2′-groups, like 2′-H, 2′-F, 2′-NH2, 2′-OMe and 2′-N3, into RNA for further application.23–26
We have tested two of those mutants for their ability to incorporate 2′-methylseleno (2′-SeCH3)-modified nucleotides into RNA. The 2′-SeCH3 group offers the possibility for application in X-ray crystallography since selenium derivatization of RNA represents a powerful method for structure determination using the multiwavelength anomalous dispersion technique (MAD).27–302′-Methylseleno-modified RNA has been successfully used in several X-ray crystallographic studies and played a crucial role in the structure determination of the Diels–Alder ribozyme.31–33 The availability of all four 2′-SeCH3 nucleoside phosphoramidite building blocks permits great flexibility for adequate positioning within the RNA target during solid phase synthesis.34–38 In addition, structural studies using the 2′-methylseleno-modified RNA target sequences showed that, for the majority of tested sequences, this type of modification did not affect the crystallization behavior or the native fold and was reported to even support the crystallization process.37,39 Another advantage of 2′-SeCH3-modified RNA is its increased resistance towards X-ray-induced radiation damage and it is thus an attractive alternative to commonly used 5-halogen-RNA.39
Currently, 2′-SeCH3-modified RNA is accessible in sizes of up to 50 nucleotides by chemical synthesis and up to about 100 nucleotides in combination with enzymatic ligation techniques.33,40 Since both of these preparation techniques are laborious and are only applicable to oligonucleotides of restricted sizes, they pose severe limitations to making this method viable in practice. For the large number of novel and highly relevant non-coding RNA sequences of 50–200 nucleotides that await structural characterization, an enzymatic approach would be of great interest to overcome these drawbacks. However, until now, no RNA polymerase has been reported that is able to incorporate 2′-methylseleno-modified nucleotides. Motivated to overcome the limitations of chemical RNA synthesis on solid supports, our efforts focused on the challenge to develop an enzymatic approach for the preparation of 2′-SeCH3-modified RNA (Fig. 1a).
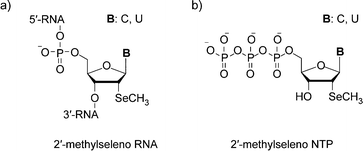 | ||
| Fig. 1 a) The chemical structure of 2′-methylseleno RNA, b) the chemical structure of the synthesized 2′-methylseleno–CTP and 2′-methylseleno–UTP. | ||
In short, we synthesized the 2′-methylseleno ribonucleoside triphosphates of cytidine and uridine (2′- SeCH3–CTP and 2′- SeCH3–UTP (Fig. 1b)). Subsequently, we tested wild-type T7 RNA polymerase, as well as two mutants, for their acceptance of the 2′-methylseleno-modified nucleotides in in vitrotranscription experiments. Those mutants, M1 (Y639F, H784A) and M2 (Y639V, H784G, E593G, V685A), have been described for their enhanced ability to incorporate 2′-OCH3- and 2′-N3-modified NTPs.25,26 Here, we found that the wild-type T7 RNA polymerase is not able to incorporate the modified nucleotides into RNA during in vitrotranscription, while both of the mutants accept 2′-SeCH3–CTP and 2′-SeCH3–UTP as substrates and efficiently incorporate them into RNA. Therefore, this study creates the foundation for 2′-SeCH3 derivatization of large-size RNA, which is of high potential for crystallographic X-ray structure analysis.
Materials and methods
Synthesis of 2′-SeCH3 pyrimidine nucleoside triphosphates 4 and 7
Enzyme preparation and in vitrotranscription assays
Results and discussion
Synthesis
To investigate the enzymatic synthesis of 2′-methylseleno RNA, we first synthesized and characterized the nucleoside triphosphate of 2′-SeCH3 uridine. The functionalized derivative 1 (Scheme 1) represented the starting material, which was readily obtained along the lines of a previously introduced synthetic route.41 Subsequent treatment with acetic anhydride resulted in quantitative acetylation of the 3′-hydroxyl group to yield compound 2, followed by selective cleavage of the dimethoxytrityl group under acidic conditions to give derivative 3. Introduction of the triphosphate moiety at the free 5′-hydroxyl group of 3 was achieved according to the Ludwig and Eckstein approach,45 using 2-chloro-4H-1,2,3-benzodioxaphosphorin-4-one, pyrophosphate and subsequent iodine oxidation. Finally, cleavage of the 3′-O-acetyl furnished the desired 2′-SeCH3 uridine derivative 4.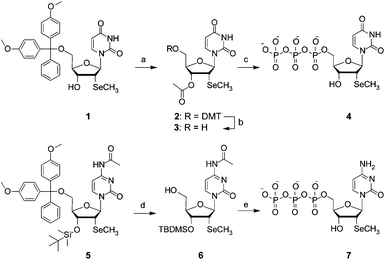 | ||
| Scheme 1 The synthesis of 2′-methylseleno uridine triphosphate 4 and 2′-methylseleno cytidine triphosphate 7. Reaction conditions: (a) 33 equiv. acetic anhydride in pyridine at room temperature for 10 h, 69%; (b) 1.2% CF3COOH in CH2Cl2 at room temperature for 15 min, 72%; (c) (i): 1.3 equiv. 2-chloro-4H-1,2,3-benzodioxaphosphorin-4-one, in dioxane/pyridine, room temperature, 10 min, (ii): 1.6 equiv. (HNBu3)2H2P2O7 in DMF and NBu3 at room temperature for 10 min, (iii): 1.2 equiv. I2 in pyridine/H2O at room temperature for 15 min, (iv): 5% (v/v) aqueous NaHSO3 solution, room temperature, 5 min, (v): 28% NH4OH solution at room temperature for 1 h, 42%; (d) 3% Cl2HCOOH in ClCH2CH2Cl at room temperature for 15 min, 81%; (e) (i): 1.3 equiv 2-chloro-4H-1,2,3-benzodioxaphosphorin-4-one in dioxane/pyridine at room temperature for 10 min, (ii): 1.6 equiv (HNBu3)2H2P2O7 in DMF and NBu3 at room temperature for 10 min, (iii): 1.2 equiv I2 in pyridine/H2O at room temperature for 15 min, (iv): 5% (v/v) aqueous NaHSO3 solution at room temperature for 5 min, (v): 28% NH4OH solution at room temperature for 1 h, 21%. | ||
Likewise, the synthesis of the 2′-SeCH3 cytidine triphosphate 7 started from the readily available 3′-O-silylated compound 5 (Scheme 1).41 The selective cleavage of the dimethoxytrityl group under acidic conditions yielded derivative 6 and this was followed by the introduction of the triphosphate moiety at the free 5′-hydroxyl group of 6, according to the Ludwig and Eckstein approach.45 Finally, cleavage of the 3′-O-TBDMS furnished the desired 2′-SeCH3 cytidine derivative 7.
Enzymes
Next, we wanted to test whether T7 RNA polymerase, an enzyme frequently used in in vitrotranscription for the synthesis of RNA, is able to accept the modified substrates. We generated T7 RNA polymerase wild-type, as well as two mutants, M1 (Y639F, H784A) and M2 (Y639V, H784G, E593G, V685A), that are known for their increased propensity to tolerate 2′-modifications.25,26 The enzymes were heterologously expressed in E. coli containing an N-terminal 6 × His-tag and purified by affinity chromatography. Interestingly, both mutants have mutations at amino acid positions Y639 and H784. The amino acid position Y639 has been implicated before in substrate recognition and an interaction with the 2′-position of incoming ribonucleotides.46,47 Both amino acid residues are located close to each other in the active site of a transcribing T7 RNA polymerase (Fig. 2).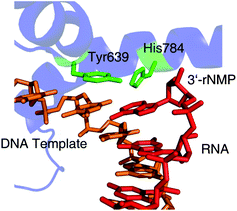 | ||
| Fig. 2 The location of Tyr639 and His784 as seen in the crystal structure of a transcribing T7 RNA polymerase (PDB 1MSW). The DNA template is shown in orange, RNA transcript in red, interesting amino acid residues in green and the RNA polymerase in blue. | ||
In vitro transcription
For in vitrotranscription using T7 RNA polymerase, a double stranded DNA template (112 nucleotides) containing the T7 promoter sequence was designed to allow the incorporation of 12 C or U residues into the RNA transcript (89 nucleotides). The RNA transcripts were labeled by the addition of [α-32P]GTP to the enzymatic reactions and the transcripts were visualized using a phosphorimager after separation by denaturating polyacrylamide gel electrophoresis (PAGE).We first tested whether the wild-type T7 RNA polymerase was able to incorporate 2′-SeCH3–CMP into an 89mer RNA transcript containing 12 C residues (Fig. 3a). PAGE analysis demonstrated that the full-length transcript could not be detected either when CTP was replaced by its 2′-methylseleno-modified analog (Fig. 3b, lane 2, WT) nor in the control experiment, where CTP was absent (Fig. 3b, lane 3, WT). We then tested the ability of the mutants M1 and M2 to incorporate 2′-SeCH3–CMP into RNA. The results indicate that both mutants are using 2′-SeCH3–CTP as a substrate and the full-length transcript is formed (Fig. 3b, lane 2, M1 and M2). In case of mutant M1, a small amount of the full-length transcript was also formed in the control reaction without any CTP, leading to the assumption that this mutant is less accurate on the provided DNA template and misincorporation is taking place (Fig. 3b, lane 3, M1). This was also observed for mutant M2, but in insignificantly low amounts (Fig. 3b, lane 3, M2). With respect to the applications for X-ray crystallography, the resulting heterogeneous mixture of transcripts could lead to difficulties during the crystallization process.
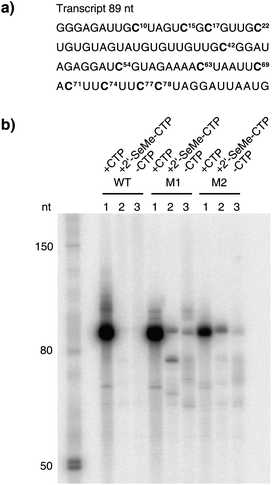 | ||
| Fig. 3 The enzymatic incorporation of 2′-SeCH3–CMP into RNA. a) The sequence of the RNA transcript (89 nt). Positions where CMP or 2′-SeCH3–CMP were incorporated into RNA are highlighted. b) The PAGE analysis of the transcription reactions comparing T7 RNA polymerase wild-type (WT) with mutant M1 and mutant M2 after 2 h of incubation at 37 °C. In each set, lane 1 shows the reaction with all four NTPs, lane 2 shows the reaction where CTP was replaced by 2′-SeCH3–CTP and lane 3 shows the control reaction without CTP but with ATP, GTP and UTP. | ||
In case of mutant M1, the polymerase discontinues transcription when incorporating two 2′-SeCH3–CMP moieties consecutively at positions 77 and 78 to form a shorter, truncated transcript (Fig. 3b, lane 2, M1). The positive control with all four NTPs demonstrated that the three tested enzymes are highly active when their natural substrates are provided (Fig. 3b, lane 1).
We performed a time-course experiment to gain insights into the incorporation efficiencies of 2′-SeCH3–CMP into RNA, comparing T7 RNA polymerase wild-type with the mutants M1 and M2. The PAGE analysis clearly indicates that 2′-SeCH3-modified RNA is formed in case of the mutants M1 and M2, but not when using the wild-type polymerase (Fig. 4a). The quantification of the full-length transcript shows that mutant M2 is approximately twice as efficient as mutant M1 when incorporating 2′-SeCH3–CMP (Fig. 4b).
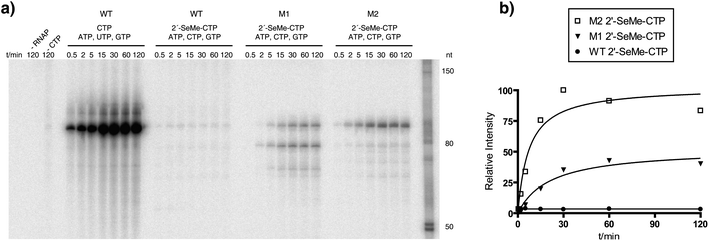 | ||
| Fig. 4 A time-course experiment for the incorporation of 2′-SeCH3–CMP into RNA. a) PAGE analysis of the transcription reactions using T7 RNA polymerase wild-type (WT), mutant M1 and mutant M2. Aliquots of the reaction mixtures were withdrawn at the corresponding time points and quenched with an equal volume of stop solution containing 50 mM EDTA. Control reactions were done with no polymerase ( −RNAP) and with no CTP but with ATP, GTP and UTP (−CTP). b) Quantification of the time-course experiment for the formation of RNA in the presence of 2′-SeCH3–CTP. | ||
Next, we determined whether 2′-SeCH3–UTP is a substrate for T7 RNA polymerase. Therefore, a DNA template calling for an 89mer transcript, where all of the C residues from our previous experiments were exchanged by U and vice versa, was used (Fig. 5a).
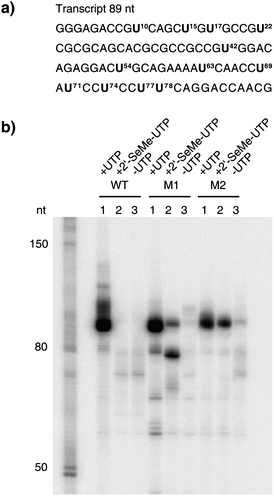 | ||
| Fig. 5 The enzymatic incorporation of 2′-SeCH3–UMP into RNA. a) The sequence of the RNA transcript (89 nt). Positions where UMP or 2′-SeCH3–UMP were incorporated into RNA are highlighted. b) PAGE analysis of the transcription reactions, comparing T7 RNA polymerase wild-type (WT) with mutant M1 and mutant M2 after 2 h of incubation at 37 °C. In each set, lane 1 shows the reaction with all four NTPs, lane 2 shows the reaction where UTP was replaced by 2′-SeCH3–UTP and lane 3 shows the control reaction without UTP but with ATP, GTP and CTP. | ||
We performed transcription reactions where UTP was replaced by its 2′-SeCH3 counterpart. In the case of the wild-type T7 RNA polymerase, no full-length transcript could be detected after PAGE analysis (Fig. 5b, lane 2, WT). For the mutants, M1 and M2, the PAGE analysis indicates that 2′-SeCH3–UMP was efficiently incorporated (Fig. 5b, lane 2, M1 and M2). No full-length transcript was detected in the control reactions without any UTP (Fig. 5b, lane 3) leading to the assumption that the formation of the full-length transcript in the reactions where 2′- SeCH3–UTP was added was due to the incorporation of 2′-SeCH3–UMP into RNA. As already seen in our previous experiments, after incorporating two 2′-SeCH3–UMP moieties consecutively at positions 77 and 78, mutant M1 was stalling, leading to a shorter transcript (Fig. 5b, lane 2, M1). The time-course experiment demonstrated that 2′-SeCH3–UMP was efficiently incorporated into RNA by mutants M1 and M2, but not when using the wild-type T7 RNA polymerase (Fig. 6a). The quantification of the full-length transcript showed that mutant M2 is most efficiently incorporating 2′-SeCH3–UMP (Fig. 6b).
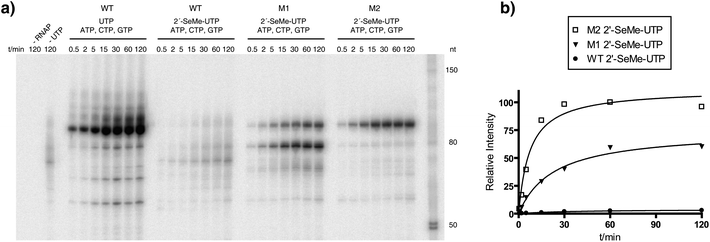 | ||
| Fig. 6 A time-course experiment for the incorporation of 2′-SeCH3–UMP into RNA. a) PAGE analysis of the transcription reactions using T7 RNA polymerase wild-type (WT), mutant M1 and mutant M2. Aliquots of the reaction mixtures were withdrawn at the corresponding time points and quenched with an equal volume of stop solution containing 50 mM EDTA. Control reactions were done with no polymerase (−RNAP) and with no UTP but with ATP, GTP and CTP (−UTP). b) Quantification of the time-course experiment for the formation of RNA in the presence of 2′-SeCH3–UTP. | ||
To determine the quantity of total RNA that can be synthesized by the T7 RNA polymerase mutant M2 in the presence of either 2′-SeCH3–UTP or UTP, we performed large scale transcription assays under the same conditions as used before and PAGE-purified the RNA transcripts. This step was necessary to remove truncated RNA molecules from the reaction mixtures that would have influenced the quantification of the desired full-length transcripts. With this procedure, it was possible to recover from a 400 μL transcription assay 90 μg (3.12 nmol) of unmodified RNA synthesized by wild-type T7 RNA polymerase, 35.3 μg (1.22 mol) of unmodified RNA synthesized by T7 RNA polymerase mutant M2 and 19.5 μg (0.65 nmol) of 2′-SeCH3-modified RNA synthesized by M2, respectively. These results are consistent with the ratios observed in the time-course experiment, where the amount of unmodified RNA generated with the wild-type enzyme was determined to be approximately six-fold higher than the amount of 2′-SeCH3-modified RNA synthesized by the mutant M2.
Conclusions
In summary, we have demonstrated the enzymatic synthesis of 2′-methylseleno-modified RNA by using the T7 RNA polymerase mutants M1 (Y639F, H784A) and M2 (Y639V, H784G, E593G, V685A) known from the literature.25,26 The experiments indicate that 2′-SeCH3–CMP and 2′-SeCH3–UMP were successfully incorporated into RNA. The best results were obtained for the incorporation of 2′-SeCH3-modified U using mutant M2. The resulting RNA is highly selenium-modified, since all 12 C or U moieties within the transcript were replaced by their corresponding 2′-SeCH3-modified counterpart. Our approach can be used, in particular, for the preparation of long 2′-SeCH3-modified RNA targets that are inaccessible by chemical solid-phase synthesis, to determine their three-dimensional structure or to study their interaction with proteins by X-ray crystallography using the multiwavelength anomalous dispersion technique (MAD). We could show that both T7 RNA polymerase mutants that have been found either by site-directed mutagenesis studies or by directed evolution for their increased acceptance of 2′-modified NTPs could also be used for the synthesis of 2′-SeCH3-modified RNA. These results are promising and a step towards an engineered RNA polymerase that processes all four 2′-SeCH3–NTPs efficiently by means of directed protein evolution.Acknowledgements
This work was funded by the DFG and by the Austrian Science Foundation FWF (I317 and P21641 to R.M.). We gratefully acknowledge support from the Konstanz Research School Chemical Biology.Notes and references
- O. Thum, S. Jager and M. Famulok, Angew. Chem., Int. Ed., 2001, 40, 3990–3993 CrossRef CAS.
- S. Jager, G. Rasched, H. Kornreich-Leshem, M. Engeser, O. Thum and M. Famulok, J. Am. Chem. Soc., 2005, 127, 15071–15082 CrossRef.
- J. P. Anderson, B. Angerer and L. A. Loeb, BioTechniques, 2005, 38, 257–264 CrossRef CAS.
- P. Capek, H. Cahova, R. Pohl, M. Hocek, C. Gloeckner and A. Marx, Chem.–Eur. J., 2007, 13, 6196–6203 CrossRef.
- T. Ohbayashi, M. Kuwahara, M. Hasegawa, T. Kasamatsu, T. Tamura and H. Sawai, Org. Biomol. Chem., 2005, 3, 2463–2468 RSC.
- G. Giller, T. Tasara, B. Angerer, K. Muhlegger, M. Amacker and H. Winter, Nucleic Acids Res., 2003, 31, 2630–2635 CrossRef CAS.
- T. Tasara, B. Angerer, M. Damond, H. Winter, S. Dorhofer, U. Hubscher and M. Amacker, Nucleic Acids Res., 2003, 31, 2636–2646 CrossRef CAS.
- P. M. Gramlich, C. T. Wirges, J. Gierlich and T. Carell, Org. Lett., 2008, 10, 249–251 CrossRef CAS.
- M. Kuwahara, J. Nagashima, M. Hasegawa, T. Tamura, R. Kitagata, K. Hanawa, S. Hososhima, T. Kasamatsu, H. Ozaki and H. Sawai, Nucleic Acids Res., 2006, 34, 5383–5394 CrossRef CAS.
- B. Seelig and A. Jaschke, Bioconjugate Chem., 1999, 10, 371–378 CrossRef CAS.
- J. Wolf, V. Dombos, B. Appel and S. Muller, Org. Biomol. Chem., 2008, 6, 899–907 RSC.
- D. Williamson, M. J. Cann and D. R. Hodgson, Chem. Commun., 2007, 5096–5098 RSC.
- R. Fiammengo, K. Musilek and A. Jaschke, J. Am. Chem. Soc., 2005, 127, 9271–9276 CrossRef CAS.
- F. Huang, J. He, Y. Zhang and Y. Guo, Nat. Protoc., 2008, 3, 1848–1861 Search PubMed.
- J. D. Vaught, T. Dewey and B. E. Eaton, J. Am. Chem. Soc., 2004, 126, 11231–11237 CrossRef CAS.
- J. F. Milligan and O. C. Uhlenbeck, Methods Enzymol., 1989, 180, 51–62 Search PubMed.
- N. Carrasco, J. Caton-Williams, G. Brandt, S. Wang and Z. Huang, Angew. Chem., Int. Ed., 2005, 45, 94–97.
- S. G. Srivatsan and Y. Tor, J. Am. Chem. Soc., 2007, 129, 2044–2053 CrossRef CAS.
- E. L. Chernolovskaya and M. A. Zenkova, Curr. Opin. Mol. Ther., 2010, 12, 158–167 Search PubMed.
- M. A. Behlke, Oligonucleotides, 2008, 18, 305–319 CrossRef CAS.
- J. K. Watts, G. F. Deleavey and M. J. Damha, Drug Discovery Today, 2008, 13, 842–855 CrossRef CAS.
- G. Mayer, Angew. Chem., Int. Ed., 2009, 48, 2672–2689 CrossRef CAS.
- R. Sousa and R. Padilla, EMBO J, 1995, 14, 4609–4621 CAS.
- R. Padilla and R. Sousa, Nucleic Acids Res., 1999, 27, 1561–1563 CrossRef CAS.
- R. Padilla and R. Sousa, Nucleic Acids Res., 2002, 30, 138e CrossRef.
- J. Chelliserrykattil and A. D. Ellington, Nat. Biotechnol., 2004, 22, 1155–1160 CrossRef CAS.
- M. Teplova, C. J. Wilds, Z. Wawrzak, V. Tereshko, Q. Du, N. Carrasco, Z. Huang and M. Egli, Biochimie, 2002, 84, 849–858 CrossRef CAS.
- N. Carrasco, D. Ginsburg, Q. Du and Z. Huang, Nucleosides, Nucleotides Nucleic Acids, 2001, 20, 1723–1734 CrossRef CAS.
- Q. Du, N. Carrasco, M. Teplova, C. J. Wilds, M. Egli and Z. Huang, J. Am. Chem. Soc., 2002, 124, 24–25 CrossRef CAS.
- N. Carrasco, Y. Buzin, E. Tyson, E. Halpert and Z. Huang, Nucleic Acids Res., 2004, 32, 1638–1646 CrossRef CAS.
- S. Freisz, K. Lang, R. Micura, P. Dumas and E. Ennifar, Angew. Chem., Int. Ed., 2008, 47, 4110–4113 CrossRef CAS.
- A. Serganov, S. Keiper, L. Malinina, V. Tereshko, E. Skripkin, C. Hobartner, A. Polonskaia, A. T. Phan, R. Wombacher, R. Micura, Z. Dauter, A. Jaschke and D. J. Patel, Nat. Struct. Mol. Biol., 2005, 12, 218–224 CrossRef CAS.
- C. Hobartner, R. Rieder, C. Kreutz, B. Puffer, K. Lang, A. Polonskaia, A. Serganov and R. Micura, J. Am. Chem. Soc., 2005, 127, 12035–12045 CrossRef.
- B. Puffer, H. Moroder, M. Aigner and R. Micura, Nucleic Acids Res., 2008, 36, 970–983 CAS.
- Y. Buzin, N. Carrasco and Z. Huang, Org. Lett., 2004, 6, 1099–1102 CrossRef CAS.
- J. Sheng, J. Jiang, J. Salon and Z. Huang, Org. Lett., 2007, 9, 749–752 CrossRef CAS.
- H. Moroder, C. Kreutz, K. Lang, A. Serganov and R. Micura, J. Am. Chem. Soc., 2006, 128, 9909–9918 CrossRef CAS.
- J. Sheng and Z. Huang, Chem. Biodiversity, 2010, 7, 753–785 CrossRef CAS.
- V. Olieric, U. Rieder, K. Lang, A. Serganov, C. Schulze-Briese, R. Micura, P. Dumas and E. Ennifar, RNA, 2009, 15, 707–715 CrossRef CAS.
- K. Lang and R. Micura, Nat. Protoc., 2008, 3, 1457–1466 Search PubMed.
- C. Hobartner and R. Micura, J. Am. Chem. Soc., 2004, 126, 1141–1149 CrossRef.
- R. Micura, C. Hobartner, R. Rieder, C. Kreutz, B. Puffer, K. Lang and H. Moroder, Curr. Protoc. Nucleic Acid Chem., 2007,(Chapter 1) Search PubMed , Unit 1 15.
- B. Puffer, H. Moroder, M. Aigner and R. Micura, Nucleic Acids Res., 2008, 36, 970–983 CAS.
- R. Peist, A. Koch, P. Bolek, S. Sewitz, T. Kolbus and W. Boos, J. Bacteriol., 1997, 179, 7679–7686 CAS.
- J. Ludwig and F. Eckstein, J. Org. Chem., 1989, 54, 631–635 CrossRef.
- Y. Huang, F. Eckstein, R. Padilla and R. Sousa, Biochemistry, 1997, 36, 8231–8242 CrossRef CAS.
- L. G. Brieba and R. Sousa, Biochemistry, 2000, 39, 919–923 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C-NMR spectra of compounds 1–7, 31P-NMR spectra of compounds 4 and 7. SDS-PAGE gel of purified T7 RNA polymerases. See DOI: 10.1039/c1sc00404b |
| This journal is © The Royal Society of Chemistry 2011 |