Synthesis of natural-product-like scaffolds in unprecedented efficiency via a 12-fold branching pathway†‡
Diane
Robbins
a,
Annabella F.
Newton
a,
Camille
Gignoux
a,
Jean-Christophe
Legeay
a,
Alex
Sinclair
b,
Martin
Rejzek
b,
Carly A.
Laxon
b,
Sai K.
Yalamanchili
b,
William
Lewis
a,
Maria A.
O'Connell
*b and
Robert A.
Stockman
*a
aSchool of Chemistry, University of Nottingham, Nottingham, UK. E-mail: robert.stockman@nottingham.ac.uk; Fax: +44 (0)115 9513654; Tel: +44 (0)115 9513252
bSchool of Pharmacy, University of East Anglia, Norwich, UK. E-mail: m.oconnell@uea.ac.uk; Tel: +44 (0)1603 592030
First published on 24th August 2011
Abstract
Tying the knot! The marriage of two-directional synthesis and tandem reactions allows access to twelve skeletally diverse scaffolds in just fifteen reactions. Two-directional synthesis yields a symmetrical linear “rope-like” keto-dienoate which is then subjected to twelve separate tandem reactions to “tie the rope in knots” thus creating twelve diverse natural product-like scaffolds containing useful functionality for further elaboration.
A key challenge in chemical biology or drug discovery is the identification of new modulators of biological systems. Traditionally new lead compounds have come from nature (natural product isolation) or from combinatorial chemistry.1 Despite a huge investment in combinatorial chemistry throughout the late 1980's and 1990's, this approach has not led to the expected surge of new chemical entities released to market.2 Much of the output of the millions of compounds made through combinatorial chemistry was compounds which were simply easy to synthesise (peptides, sulfonamides or simple aromatic compounds) with little thought for how “drug-like” they might be.3 It became clear that for a successful screening campaign, the library of compounds needs to be diverse in three-dimensional shape, stereochemistry and functionality,4 and it need not necessarily have to contain thousands of compounds, but should be diverse in its coverage of “chemical space.”5 Diversity-Oriented-Synthesis (DOS) aims to meet this challenge by creating complexity from simple starting materials to generate libraries of diverse complex molecules.6 Several strategies for DOS have emerged since Schreiber coined the term in the late 1990's.7 Despite over a decade passing, the rapid access to a wide range of three-dimensional compounds with useful functionalities remains a formidable challenge for DOS.8,9 The needs of chemical biology/chemical genetics and the pharmaceutical industry in terms of diverse compound libraries are starting to overlap more and more. Three-dimensional compounds are having a renaissance in the pharmaceutical industry, as it has been noted that often compounds which have more three-dimensionality are less liable to fail on the long trail to becoming an approved API.10 Current attrition rates of compounds reaching the clinic are 97%, and thus industry now perceives requirements of potential lead compounds to not only satisfy the Lipinski “rule of 5,”11 but also have an increased number of sp3 atoms, more polar functionality, and smaller molecular weight.12 Thus one could suggest that this new “Lead-Oriented-Synthesis”13 is a subset of DOS, which was originally conceived for chemical genetics,14 where the function of the molecule is much more important than bioavailability, and thus size and lipophilicity of the library is not such a constraint. Furthermore, taking a lead from natural products, which have a large structural diversity,15 the synthesis of “natural product-like” libraries,16 and “biologically-oriented-synthesis”17 aim to exploit some of the lessons we can learn from natural products which have been “pre-validated” through evolution, and couple this with the exploration of chemical space paradigm of DOS.18 Indeed in a recent screen against a panel of 100 proteins, Schreiber found that a collection of DOS-inspired compounds offered better “hit-rates” and selectivity over both a collection of natural products and a collection of commercial compounds.19
Over the past decade we have been interested in the idea of combining two-directional synthesis with tandem reactions to yield complex molecules in very short order.20 This strategy has yielded some extremely short syntheses of natural products from our labs.21 More recently we became curious as to whether this approach may also be a powerful strategy for DOS. Much like one can tie a number of different knots in a piece of rope to yield a variety of different three-dimensional shapes, we wondered if we could apply a range of tandem or cascade reactions to “tie” the linear “rope-like” molecules furnished by two-directional synthesis into a multitude of three-dimensional molecular architectures, laden with stereogenic centres, a variety of ring systems and heteroatoms and with useful functionality to further diversify with more standard “capping” type transformations.22 Spurred on by the use of the substrate 1 in both the synthesis of the pinnaic acid core structure 2,23 in a unique 8-step cascade transformation24 to yield pyrrolidin-dione 3, and in the synthesis of the beetle kairomone hippodamine (via4),25 we decided to investigate and invent more methods of tying this molecule into three-dimensional scaffolds (Scheme 1). The molecule 1 is well suited to an investigation of a diversity-approach based on tandem reactions due to its very simple three-step synthesis26 and its functionality: a central ketone is able to be transformed into a range of nucleophilic functionalities, which can then react with the tethered α,β-unsaturated esters at the termini of the chain. Moreover, we surmised that if we can develop tandem reactions which allow the central functionality to react with one chain-end, transforming the central functionality, and subsequently react with the other chain-end under a different mode of reactivity, then we would be able to create a non-symmetrical three-dimensional molecule from the simple linear substrate. An exemplar of this desymmetrising or transformative tandem reaction concept is shown in Scheme 2.
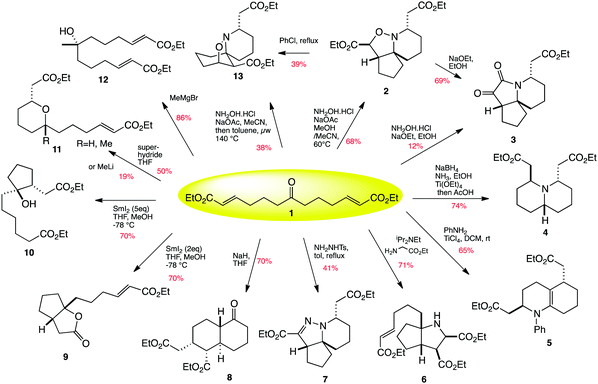 | ||
| Scheme 1 Twelve-fold branching synthesis. | ||
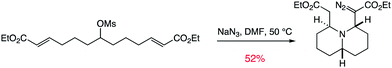 | ||
| Scheme 2 An example of a transformative tandem reaction. | ||
Having already looked at the transformation of the central ketone functionality of keto-diester 1 into an oxime, followed by a tandem aza-Michael/1,4-prototopic shift/intramolecular [3 + 2] cycloaddition which yielded azaspirocycle 2 in 68% yield as a single diastereomer (Scheme 1),23 we initially turned our attention to other amine-based transformations of the central ketone of 1. Thus reaction of compound 1 with aniline in the presence of the strong Lewis-acid titanium(IV) chloride induced an enamine formation/Michael/aza-Michael cascade which gave octahydroquinoline 5 in 65% yield as a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of trans to cis diastereomers (Scheme 1). There is a relatively low energy difference between the two diastereomers formed, and thus it is assumed that the product is formed as an equilibrium mixture of these two diastereomers.
:1 mixture of trans to cis diastereomers (Scheme 1). There is a relatively low energy difference between the two diastereomers formed, and thus it is assumed that the product is formed as an equilibrium mixture of these two diastereomers.
With this initial success, further amine-induced transformations of the central ketone were investigated. Thus treatment of ketone 1 with glycine methyl ester in the presence of Hünigs base induced a condensation/azomethine ylid formation/[3 + 2] cycloaddition to yield the octahydrocyclopenta[b]pyrrole 6 in 71% yield, again as a single diastereomer (Scheme 1). Further potential aza-Michael addition is not seen, presumably due to steric factors.
Having developed cascades involving nitrone and azomethine ylide formation and cycloaddition, we surmised that we should be able to induce a transformation based on an azomethine imine cycloaddition. Thus reaction of ketone 1 with tosyl hydrazone in the presence of Hünigs base did indeed promote a cascade of hydrazone formation/aza-Michael/prototopic shift/[3 + 2] cycloaddition, but to our mild surprise a further elimination event completed the sequence to yield cyclic hydrazone 7 as a single diastereomer (Scheme 1).
The central ketone of 1 itself can of course be used as a pro-nucleophilic functionality. Thus treatment of ketone 1 with sodium hydride gives an enolate which is able to carry out a Michael addition with one end of the chain to give an ester enolate, which further cyclises through another Michael addition to the other end of the chain to give keto decalin 8 as a single diastereomer (Scheme 3). We were fortunate enough to crystallise this material to confirm that this remarkable transformation had yielded a bicyclic product with four contiguous stereogenic centres in a single step and with complete diastereocontrol!
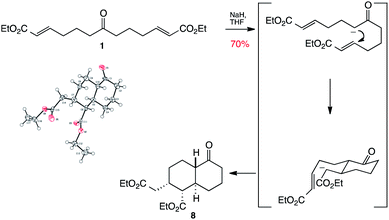 | ||
| Scheme 3 Synthesis and X-ray structure of decalin 8. | ||
Ketones can also be reduced to ketyl radicals, which we thought would offer interesting ambident reactivity towards the α,β-unsaturated side-chains. It was found that on treating ketone 1 with two equivalents of samarium(II) iodide, a transformation resulting in the bicyclic lactone 9 was observed. If five equivalents of samarium iodide were used, a divergence in the stereochemical outcome of the presumed carbon radical cyclisation onto the first alkene was noted, with the excess reducing agent in this case reducing the resultant ester-stabilised radical to an enolate and protonation by methanol and indeed reduction of the other alkene to the alkane, yielding carbocycle 10. Presumably in the case where an excess of samarium iodide is used, two separate samarium molecules can be involved in the initial radical cyclisation, leading to the trans cyclised product by steric hindrance, and quickly reducing the ester-stabilised radical to the enolate. In the case where only two equivalents of SmI2 are used, the first equivalent again makes the ketyl radical, with the alkoxide anion bound to a Sm(III). This Sm(III) can internally chelate an ester group of the pendant alkene, controlling the attack of the radical onto the alkene to form a ciscyclopentane. The alkoxide can then attack the proximal ester to form a lactone.
Ketones can of course react with nucleophiles, and thus we investigated the reaction of ketone 1 with Grignard reagents and with hydride sources such as superhydride. It was found that Grignard addition led only to the formation of a symmetrical tertiary alcohol (12Scheme 1), whereas reaction with superhydride reduced the ketone to an alkoxide, which then underwent subsequent oxy-Michael addition to yield 2,6-tetrahydropyran 11a (R = H) as a single diastereomer. Methyl lithium also followed the same type of addition/cyclisation to yield 11b (R = Me) in low yield, with the majority of the reaction taking the pathway previously observed with sodium hydride, yielding decalin 8 (Scheme 1).
In our previous work towards pinnaic acid, we found that 1 could be transformed into the [5.5.6] spirocycle 2, and that this kinetic product could subsequently be converted into the thermodynamic regioisomer 13 by heating in chlorobenzene (Scheme 4).22 We have now found that heating 1 with sodium acetate and hydroxylamine hydrochloride in methanol and acetonitrile at elevated temperatures of 140 °C in a microwave is able to render the thermodynamic product 13 directly accessible from 1, thus making this interesting transformation capable of generating two distinct and useful tricyclic skeletons depending on the temperature used.
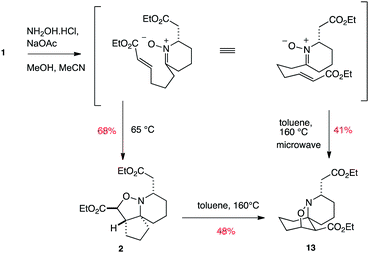 | ||
| Scheme 4 | ||
As an exemplar of this approach to the synthesis of scaffolds suitable for finding new molecules with interesting biological activity, we synthesised a small library of analogues from scaffold 2, and tested them against three cancer cell lines.27 All compounds were available in 6 steps or less from 2, with 17 reactions in total being used to produce the 8 analogues tested. The results of this screen are shown in Table 1. As would be expected, a range of activities are noted, with compound 17 being the most active for all three cell lines, and others showing selectivity (e.g. removal of the hydroxyl group of 17 to give 18 removes activity from the A549 cell line).28 For comparison we tested doxorubicin in the HL-60 assay, which gave a result of 0.02 μM. Whilst this is not a medicinal chemistry study, these results do confirm that this is a viable methodology for the rapid access of three-dimensionally shaped scaffolds that are capable of being diversified themselves into a range of molecules suitable for biological screening, and which can give activities appropriate for further investigation as potential leads for a medicinal chemistry programme or as potential biological probes for chemical biology.
| Compound | IC50 values/μM | ||
|---|---|---|---|
| HL-60 | THP-1 | A549 | |

|
— | >100 | — |

|
22.4 | 27.5 | 41.3 |
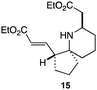
|
>100 | >100 | >100 |
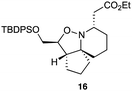
|
24 | 33 | 29 |
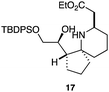
|
4.7 | 2.1 | 22.4 |
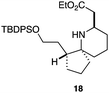
|
11.0 | 3.5 | >100 |
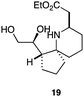
|
48.8 | >100 | >100 |
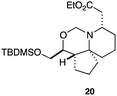
|
34 | 44 | 44 |
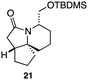
|
42 | 36 | 43 |
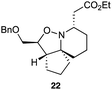
|
18.6 | >100 | — |
In summary, we have demonstrated the concept of formation of a simple linear “molecular rope” and using tandem reactions to “tie it into knots” as an interesting and very direct approach for the efficient formation of a range of three-dimensional molecular architectures with natural product-like features (polycyclic ring systems, multiple stereocentres, range of functional groups). This approach is able to access a range of carbocycles, azacycles and oxocycles, with fused, spiro and bridged polycyclic structures from a single substrate. Thus a large number of interesting and useful scaffolds have been delivered in an amazing 1.25 steps per new scaffold on average. Studies using a similar approach with other substrates are ongoing and will be reported in due course.
Acknowledgements
The authors gratefully acknowledge funding from the University of Nottingham (DR, CG), University of East Anglia (MOC, CAL), Leverhulme Trust (MR), Novartis Summer Studentship Programme (SKY) and EPSRC (Advanced Research Fellowship EP/E055346/1, RAS, AFN, JCL, AS).Notes and references
- (a) G. M. Cragg, P. G. Grothaus and D. L. Newman, Chem. Rev., 2009, 109, 3012–3043 CrossRef CAS; (b) D. J. Newman and G. M. Cragg, J. Nat. Prod., 2007, 70, 461–477 CrossRef CAS.
- D. J. Newman, J. Med. Chem., 2008, 51, 2589–2599 CrossRef CAS.
- S. J. Teague, A. M. Davis, P. D. Leeson and T. Oprea, Angew. Chem., Int. Ed., 1999, 38, 3743–3748 CrossRef CAS.
- M. M. Hann, A. R. Leach and G. Harper, J. Chem. Inf. Comput. Sci., 2001, 41, 856–864 CrossRef CAS.
- M. K. Schwartz and W. H. B. Sauer, J. Chem. Inf. Comput. Sci., 2003, 43, 987–1003 CrossRef CAS.
- M. D. Burke and S. L. Schreiber, Angew. Chem., Int. Ed., 2004, 43, 46–58 CrossRef.
- R. J. Spandl, A. Bender and D. R. Spring, Org. Biomol. Chem., 2008, 6, 1149–1158 RSC.
- M. D. Burke, E. M. Berger and S. L. Schreiber, Science, 2003, 302, 613–618 CrossRef CAS.
- W. R. J. D. Galloway and D. R. Spring, Expert Opin. Drug Discovery, 2009, 4, 467–472 Search PubMed.
- F. Lovering, J. Bikker and C. Humbler, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef.
- T. E. Nielsen and S. L. Schreiber, Angew. Chem., Int. Ed., 2008, 47, 48–56 CrossRef CAS.
- The phrase “Lead-Oriented Synthesis” was used by Drs Ian Churcher and Alan Nadin at the GSK Novel Synthetic Methods Symposium, GSK Stevenage, 24–25 May 2010. To the authors’ knowledge, this is its first use.
- S. L. Schreiber, Science, 2000, 287, 1964–1969 CrossRef CAS.
- M. A. Koch, A. Schuffenhauer, M. Scheck, S. Wetzel, M. Casaulta, A. Odermatt, P. Ertl and H. Waldmann, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17272–17277 CrossRef CAS.
- D. Morton, S. Leach, C. Cordier, S. Warriner and A. Nelson, Angew. Chem., Int. Ed., 2008, 48, 104–109 CrossRef.
- A. Nören-Müller, I. Reis-Corrêa, Jr, H. Prinz, C. Rosenbaum, K. Saxena, H. J. Schwalbe, D. Vestweber, G. Cagna, S. Schunk, O. Schwarz, H. Schiewe and H. Waldmann, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10606–10611 CrossRef.
- K. Kumar and H. Waldmann, Angew. Chem., Int. Ed., 2009, 48, 1740–1752 CrossRef CAS.
- P. A. Clemons, N. E. Bodycombe, H. A. Carrinski, J. A. Wilson, A. F. Shamji, B. K. Wagner, A. N. Koehler and S. L. Schreiber, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18787–18792 CrossRef CAS.
- For a recent example, see: J.-C. Legeay, W. Lewis and R. A. Stockman, Chem. Commun., 2009, 2207–2209 Search PubMed.
- For example: M. S. Karatholuvhu, A. Sinclair, A. F. Newton, M.-L. Alcaraz, R. A. Stockman and P. L. Fuchs, J. Am. Chem. Soc., 2006, 128, 12656–12657 Search PubMed.
- An example of a desymmetrising tandem reaction: M. Rejzek, R. A. Stockman, J. H. van Maarseveen and D. L. Hughes, Chem. Commun., 2005, 4661–4662 Search PubMed.
- L. G. Arini, P. Szeto, D. L. Hughes and R. A. Stockman, Tetrahedron Lett., 2004, 45, 8371–8374 CrossRef CAS.
- A. Sinclair, L. G. Arini, M. Rejzek, P. Szeto and R. A. Stockman, Synlett, 2006, 2321–2324 CAS.
- A. F. Newton, M. Rejzek, M.-L. Alcaraz and R. A. Stockman, Beilstein J. Org. Chem., 2008, 4, 4 Search PubMed.
- A. F. Newton, S. J. Roe, J.-C. Legeay, P. Aggarwal, C. Gignoux, N. J. Birch, R. Nixon, M.-L. Alcaraz and R. A. Stockman, Org. Biomol. Chem., 2009, 7, 2274–2277 RSC.
- We chose these cell lines as similar spirocyclic piperidines were shown to be active in THP-1 assays by Itoh et al.: M. Itoh, J. Kuwahara, K. Itoh, Y. Fukuka, M. Kohya, M. Shindo and K. Shishido, Bioorg. Med. Chem. Lett., 2002, 12, 2069 Search PubMed.
- Itoh et al.27 also found compounds with silyl groups gave the highest activity in their THP-1 assays. This may be down to better cell membrane permeation of these more lipophilic compounds, although the inactivity of 18 in the A549 cell lines rules out any generic cytotoxicity effect associated with lipophilic molecules.
Footnotes |
| † During the period in which this manuscript was in review, a paper was published in which 12 scaffolds were synthesised from a single starting molecule using tandem reactions: W. Liu, V. Khedkar, B. Baskar, M. Shürmann and K. Kumar, Angew. Chem. Int. Ed., 2011, 50, 6900. |
| ‡ Electronic supplementary information (ESI) available: Postulated mechanisms, screening assay details, synthetic procedures and NMR spectra. CCDC reference number 839592. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00371b |
| This journal is © The Royal Society of Chemistry 2011 |